Functional Domain Motions and Processivity in Bacterial Hyaluronate Lyase: a Molecular Dynamics Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supporting Information High-Throughput Virtual Screening
Supporting Information High-Throughput Virtual Screening of Proteins using GRID Molecular Interaction Fields Simone Sciabola, Robert V. Stanton, James E. Mills, Maria M. Flocco, Massimo Baroni, Gabriele Cruciani, Francesca Perruccio and Jonathan S. Mason Contents Table S1 S2-S21 Figure S1 S22 * To whom correspondence should be addressed: Simone Sciabola, Pfizer Research Technology Center, Cambridge, 02139 MA, USA Phone: +1-617-551-3327; Fax: +1-617-551-3117; E-mail: [email protected] S1 Table S1. Description of the 990 proteins used as decoy for the Protein Virtual Screening analysis. PDB ID Protein family Molecule Res. (Å) 1n24 ISOMERASE (+)-BORNYL DIPHOSPHATE SYNTHASE 2.3 1g4h HYDROLASE 1,3,4,6-TETRACHLORO-1,4-CYCLOHEXADIENE HYDROLASE 1.8 1cel HYDROLASE(O-GLYCOSYL) 1,4-BETA-D-GLUCAN CELLOBIOHYDROLASE I 1.8 1vyf TRANSPORT PROTEIN 14 KDA FATTY ACID BINDING PROTEIN 1.85 1o9f PROTEIN-BINDING 14-3-3-LIKE PROTEIN C 2.7 1t1s OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.4 1t1r OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.3 1q0q OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 1.9 1jcy LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.9 1fww LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.85 1uk7 HYDROLASE 2-HYDROXY-6-OXO-7-METHYLOCTA-2,4-DIENOATE 1.7 1v11 OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.95 1x7w OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.73 1d0l TRANSFERASE 35KD SOLUBLE LYTIC TRANSGLYCOSYLASE 1.97 2bt4 LYASE 3-DEHYDROQUINATE DEHYDRATASE -
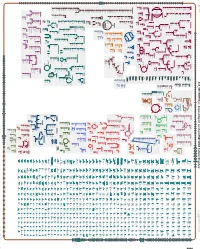
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

The Crystal Structure of Novel Chondroitin Lyase ODV-E66, A
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 587 (2013) 3943–3948 journal homepage: www.FEBSLetters.org The crystal structure of novel chondroitin lyase ODV-E66, a baculovirus envelope protein ⇑ Yoshirou Kawaguchi a, Nobuo Sugiura b, Koji Kimata c, Makoto Kimura a,d, Yoshimitsu Kakuta a,d, a Laboratory of Structural Biology, Graduate School of System Life Sciences, Kyushu University, 6-10-1 Hakozaki, Fukuoka 812-8581, Japan b Institute for Molecular Science of Medicine, Aichi Medical University, 1-1 Yazakokarimata, Nagakute, Aichi 480-1195, Japan c Research Complex for the Medicine Frontiers, Aichi Medical University, 1-1 Yazakokarimata, Nagakute, Aichi 480-1195, Japan d Faculty of Agriculture, Kyushu University, 6-10-1 Hakozaki, Fukuoka 812-8581, Japan article info abstract Article history: Chondroitin lyases have been known as pathogenic bacterial enzymes that degrade chondroitin. Received 5 August 2013 Recently, baculovirus envelope protein ODV-E66 was identified as the first reported viral chondroi- Revised 1 October 2013 tin lyase. ODV-E66 has low sequence identity with bacterial lyases at <12%, and unique characteris- Accepted 15 October 2013 tics reflecting the life cycle of baculovirus. To understand ODV-E66’s structural basis, the crystal Available online 26 October 2013 structure was determined and it was found that the structural fold resembled that of polysaccharide Edited by Christian Griesinger lyase 8 proteins and that the catalytic residues were also conserved. This structure enabled discus- sion of the unique substrate specificity and the stability of ODV-E66 as well as the host specificity of baculovirus. -

Manual D'estil Per a Les Ciències De Laboratori Clínic
MANUAL D’ESTIL PER A LES CIÈNCIES DE LABORATORI CLÍNIC Segona edició Preparada per: XAVIER FUENTES I ARDERIU JAUME MIRÓ I BALAGUÉ JOAN NICOLAU I COSTA Barcelona, 14 d’octubre de 2011 1 Índex Pròleg Introducció 1 Criteris generals de redacció 1.1 Llenguatge no discriminatori per raó de sexe 1.2 Llenguatge no discriminatori per raó de titulació o d’àmbit professional 1.3 Llenguatge no discriminatori per raó d'ètnia 2 Criteris gramaticals 2.1 Criteris sintàctics 2.1.1 Les conjuncions 2.2 Criteris morfològics 2.2.1 Els articles 2.2.2 Els pronoms 2.2.3 Els noms comuns 2.2.4 Els noms propis 2.2.4.1 Els antropònims 2.2.4.2 Els noms de les espècies biològiques 2.2.4.3 Els topònims 2.2.4.4 Les marques registrades i els noms comercials 2.2.5 Els adjectius 2.2.6 El nombre 2.2.7 El gènere 2.2.8 Els verbs 2.2.8.1 Les formes perifràstiques 2.2.8.2 L’ús dels infinitius ser i ésser 2.2.8.3 Els verbs fer, realitzar i efectuar 2.2.8.4 Les formes i l’ús del gerundi 2.2.8.5 L'ús del verb haver 2.2.8.6 Els verbs haver i caldre 2.2.8.7 La forma es i se davant dels verbs 2.2.9 Els adverbis 2.2.10 Les locucions 2.2.11 Les preposicions 2.2.12 Els prefixos 2.2.13 Els sufixos 2.2.14 Els signes de puntuació i altres signes ortogràfics auxiliars 2.2.14.1 La coma 2.2.14.2 El punt i coma 2.2.14.3 El punt 2.2.14.4 Els dos punts 2.2.14.5 Els punts suspensius 2.2.14.6 El guionet 2.2.14.7 El guió 2.2.14.8 El punt i guió 2.2.14.9 L’apòstrof 2.2.14.10 L’interrogant 2 2.2.14.11 L’exclamació 2.2.14.12 Les cometes 2.2.14.13 Els parèntesis 2.2.14.14 Els claudàtors 2.2.14.15 -

Active Site of Chondroitin AC Lyase Revealed by the Structure Of
Biochemistry 2001, 40, 2359-2372 2359 Active Site of Chondroitin AC Lyase Revealed by the Structure of Enzyme-Oligosaccharide Complexes and Mutagenesis†,‡ Weijun Huang,§ Lorena Boju,§ Lydia Tkalec,|,⊥ Hongsheng Su,|,# Hyun-Ok Yang,3 Nur Sibel Gunay,3 Robert J. Linhardt,3 Yeong Shik Kim,O Allan Matte,§ and Miroslaw Cygler*,§ Biotechnology Research Institute, 6100 Royalmount AVenue, Montre´al, Que´bec H4P 2R2 Canada, Montreal Joint Centre for Structural Biology, Montre´al, Que´bec, Canada, IBEX Technologies Inc., 5485 Pare Street, Montre´al, Que´bec H4P 1P7 Canada, Department of Chemistry, DiVision of Medicinal Chemistry and Department of Chemical and Biochemical Engineering, The UniVersity of Iowa, 115 South Grand AVenue, PHAR S328, Iowa City, Iowa 52242-1112, and Natural Products Research Institute, Seoul National UniVersity, Seoul 110-460, Korea ReceiVed October 17, 2000; ReVised Manuscript ReceiVed December 18, 2000 ABSTRACT: The crystal structures of FlaVobacterium heparinium chondroitin AC lyase (chondroitinase AC; EC 4.2.2.5) bound to dermatan sulfate hexasaccharide (DShexa), tetrasaccharide (DStetra), and hyaluronic acid tetrasaccharide (HAtetra) have been refined at 2.0, 2.0, and 2.1 Å resolution, respectively. The structure of the Tyr234Phe mutant of AC lyase bound to a chondroitin sulfate tetrasaccharide (CStetra) has also been determined to 2.3 Å resolution. For each of these complexes, four (DShexa and CStetra) or two (DStetra and HAtetra) ordered sugars are visible in electron density maps. The lyase AC DShexa and CStetra complexes reveal binding at four subsites, -2, -1, +1, and +2, within a narrow and shallow protein channel. We suggest that subsites -2 and -1 together represent the substrate recognition area, +1 is the catalytic subsite and +1 and +2 together represent the product release area. -

Wo 2008/127291 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date PCT (10) International Publication Number 23 October 2008 (23.10.2008) WO 2008/127291 A2 (51) International Patent Classification: Jeffrey, J. [US/US]; 106 Glenview Drive, Los Alamos, GOlN 33/53 (2006.01) GOlN 33/68 (2006.01) NM 87544 (US). HARRIS, Michael, N. [US/US]; 295 GOlN 21/76 (2006.01) GOlN 23/223 (2006.01) Kilby Avenue, Los Alamos, NM 87544 (US). BURRELL, Anthony, K. [NZ/US]; 2431 Canyon Glen, Los Alamos, (21) International Application Number: NM 87544 (US). PCT/US2007/021888 (74) Agents: COTTRELL, Bruce, H. et al.; Los Alamos (22) International Filing Date: 10 October 2007 (10.10.2007) National Laboratory, LGTP, MS A187, Los Alamos, NM 87545 (US). (25) Filing Language: English (81) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of national protection available): AE, AG, AL, AM, AT,AU, AZ, BA, BB, BG, BH, BR, BW, BY,BZ, CA, CH, (30) Priority Data: CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, 60/850,594 10 October 2006 (10.10.2006) US ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, (71) Applicants (for all designated States except US): LOS LR, LS, LT, LU, LY,MA, MD, ME, MG, MK, MN, MW, ALAMOS NATIONAL SECURITY,LLC [US/US]; Los MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, Alamos National Laboratory, Lc/ip, Ms A187, Los Alamos, PT, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, SV, SY, NM 87545 (US). -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Supplementary Materials
Supplementary Materials OH 8 H H8 H C N OH 3 7 3 2 4 H1 1 5 O H4 O 6 H6’ H3 OH H6 H1’ H2 H5 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 ppm 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 ppm Figure S1. 1D 1H spectrum of 1-dglcnac and signal assignments ppm 2.0 H8 2.5 OH 8 H H C N OH 3 7 3 2 4 3.0 1 5 H1 O H5 O 6 H4 H3 OH 3.5 H6 H2 H6’ H1’ 4.0 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 2.0 ppm Figure S2. 1H-1H TOCSY 2D NMR spectra for 1-dglcnac and signal assignments ppm 3.0 H1 3.2 H5 H4 3.4 H3 3.6 H6 H6’ H2 3.8 H1’ 4.0 4.1 4.0 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 3.1 3.0 ppm Figure S3. 1H-1H COSY 2D NMR spectra for 1-dglcnac and signal assignment ppm -0.02 -0.01 0.00 0.01 0.02 H1’ H6’ H2 H6 H3 H4 H5 H1 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 3.1 ppm Figure S4. 1H-1H JRES 2D NMR spectra for 1-dglcnac and signal assignments ppm 20 C8(22.9) 40 OH 8 H H C N OH C2(53.2) 3 7 3 2 4 60 1 5 C6(63.2) C6(63.2) O 6 C1(69.2) C1(69.2) O C4(72.6) C3(77.0) OH 80 C5(82.7) 100 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 2.0 ppm Figure S5. -

POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii
POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii http://rcin.org.pl WSKAZÓWKI DLA AUTORÓW Kwartalnik „Postępy Biochemii” publikuje artykuły monograficzne omawiające wąskie tematy, oraz artykuły przeglądowe referujące szersze zagadnienia z biochemii i nauk pokrewnych. Artykuły pierwszego typu winny w sposób syntetyczny omawiać wybrany temat na podstawie możliwie pełnego piśmiennictwa z kilku ostatnich lat, a artykuły drugiego typu na podstawie piśmiennictwa z ostatnich dwu lat. Objętość takich artykułów nie powinna przekraczać 25 stron maszynopisu (nie licząc ilustracji i piśmiennictwa). Kwartalnik publikuje także artykuły typu minireviews, do 10 stron maszynopisu, z dziedziny zainteresowań autora, opracowane na podstawie najnow szego piśmiennictwa, wystarczającego dla zilustrowania problemu. Ponadto kwartalnik publikuje krótkie noty, do 5 stron maszynopisu, informujące o nowych, interesujących osiągnięciach biochemii i nauk pokrewnych, oraz noty przybliżające historię badań w zakresie różnych dziedzin biochemii. Przekazanie artykułu do Redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii”. Autorzy artykułu odpowiadają za prawidłowość i ścisłość podanych informacji. Autorów obowiązuje korekta autorska. Koszty zmian tekstu w korekcie (poza poprawieniem błędów drukarskich) ponoszą autorzy. Artykuły honoruje się według obowiązujących stawek. Autorzy otrzymują bezpłatnie 25 odbitek swego artykułu; zamówienia na dodatkowe odbitki (płatne) należy zgłosić pisemnie odsyłając pracę po korekcie autorskiej. Redakcja prosi autorów o przestrzeganie następujących wskazówek: Forma maszynopisu: maszynopis pracy i wszelkie załączniki należy nadsyłać w dwu egzem plarzach. Maszynopis powinien być napisany jednostronnie, z podwójną interlinią, z marginesem ok. 4 cm po lewej i ok. 1 cm po prawej stronie; nie może zawierać więcej niż 60 znaków w jednym wierszu nie więcej niż 30 wierszy na stronie zgodnie z Normą Polską. -

Discovery of High Affinity Receptors for Dityrosine Through Inverse Virtual Screening and Docking and Molecular Dynamics
Article Discovery of High Affinity Receptors for Dityrosine through Inverse Virtual Screening and Docking and Molecular Dynamics Fangfang Wang 1,*,†, Wei Yang 2,3,† and Xiaojun Hu 1,* 1 School of Life Science, Linyi University, Linyi 276000, China; [email protected] 2 Department of Microbiology, Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia, [email protected] 3 Arieh Warshel Institute of Computational Biology, the Chinese University of Hong Kong, 2001 Longxiang Road, Longgang District, Shenzhen 518000, China * Corresponding author: [email protected] † These authors contributed equally to this work. Received: 09 December 2018; Accepted: 23 December 2018; Published: date Table S1. Docking affinity scores for cis-dityrosine binding to binding proteins. Target name PDB/UniProtKB Type Affinity (kcal/mol) Galectin-1 1A78/P56217 Lectin -6.2±0.0 Annexin III 1AXN/P12429 Calcium/phospholipid Binding Protein -7.5±0.0 Calmodulin 1CTR/P62158 Calcium Binding Protein -5.8±0.0 Seminal Plasma Protein Pdc-109 1H8P/P02784 Phosphorylcholine Binding Protein -6.6±0.0 Annexin V 1HAK/P08758 Calcium/phospholipid Binding -7.4±0.0 Alpha 1 antitrypsin 1HP7/P01009 Protein Binding -7.6±0.0 Histidine-Binding Protein 1HSL/P0AEU0 Binding Protein -6.3±0.0 Intestinal Fatty Acid Binding Protein 1ICN/P02693 Binding Protein(fatty Acid) -9.1±0.0* Migration Inhibitory Factor-Related Protein 14 1IRJ/P06702 Metal Binding Protein -7.0±0.0 Lysine-, Arginine-, Ornithine-Binding Protein 1LST/P02911 Amino Acid Binding Protein -6.5±0.0 -

Similar Structures to the E-To-H Helix Unit in the Globin-Like Fold Are Found in Other Helical Folds
Biomolecules 2014, 4, 268-288; doi:10.3390/biom4010268 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Article Similar Structures to the E-to-H Helix Unit in the Globin-Like Fold are Found in Other Helical Folds Masanari Matsuoka 1,2, Aoi Fujita 1, Yosuke Kawai 1,† and Takeshi Kikuchi 1,* 1 Department of Bioinformatics, College of Life Sciences, Ritsumeikan University, Kusatsu, Shiga 525-8577, Japan; E-Mails: [email protected] (M.M.); [email protected] (A.F.); [email protected] (Y.K.) 2 Japan Society for the Promotion of Science (JSPS), Ichibancho, Chiyoda-ku, Tokyo 102-8471, Japan † Present address: Division of Biomedical Information Analysis, Department of Integrative Genomics, Tohoku Medical Megabank Organization, Tohoku University, Sendai, Miyagi 980-8575, Japan * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +81-77-561-5909; Fax: +81-77-561-5203. Received: 6 December 2013; in revised form: 11 February 2014 / Accepted: 13 February 2014 / Published: 27 February 2014 Abstract: A protein in the globin-like fold contains six alpha-helices, A, B, E, F, G and H. Among them, the E-to-H helix unit (E, F, G and H helices) forms a compact structure. In this study, we searched similar structures to the E-to-H helix of leghomoglobin in the whole protein structure space using the Dali program. Several similar structures were found in other helical folds, such as KaiA/RbsU domain and Type III secretion system domain. These observations suggest that the E-to-H helix unit may be a common subunit in the whole protein 3D structure space. -
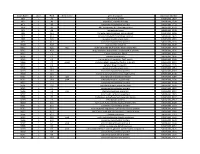
NTML 384-Well Array Well Identification
Strain Name plate Well Gene name gene discription Accession Number NE1 1 A1 cell surface protein SAUSA300_1327 NE2 1 A3 peptidase, rhomboid family SAUSA300_1509 NE3 1 A5 conserved hypothetical protein SAUSA300_1733 NE4 1 A7 ABC transporter ATP-binding protein SAUSA300_0309 NE5 1 A9 aminotransferase SAUSA300_2539 NE6 1 A11 formate dehydrogenase, alpha subunit SAUSA300_2258 NE7 1 A13 conserved hypothetical phage protein SAUSA300_1425 NE8 1 A15 putative membrane protein SAUSA300_1481 NE9 1 A17 conserved hypothetical protein SAUSA300_1294 NE10 1 A19 putative hemolysin III SAUSA300_2129 NE11 1 A21 recJ single-stranded-DNA-specific exonuclease RecJ SAUSA300_1592 NE12 1 A23 drug resistance transporter, EmrB/QacA subfamily SAUSA300_2126 NE13 1 C1 ribose transporter RbsU SAUSA300_0264 NE14 1 C3 putative transporter SAUSA300_2406 NE15 1 C5 transcriptional regulator, TetR family SAUSA300_2509 NE16 1 C7 moaD molybdopterin converting factor, subunit 1 SAUSA300_2221 NE17 1 C9 putative drug transporter SAUSA300_1705 NE18 1 C11 putative membrane protein SAUSA300_1851 NE19 1 C13 conserved hypothetical protein SAUSA300_0369 NE20 1 C15 transcriptional antiterminator, BglG family SAUSA300_0238 NE21 1 C17 upp uracil phosphoribosyltransferase SAUSA300_2066 NE22 1 C19 polA DNA polymerase I superfamily SAUSA300_1636 NE23 1 C21 conserved hypothetical protein SAUSA300_0740 NE24 1 C23 secretory antigen precursor SsaA SAUSA300_2503 NE25 1 E1 conserved hypothetical protein SAUSA300_1905 NE26 1 E3 coa staphylocoagulase precursor SAUSA300_0224 NE27 1 E5 nitric oxide