Similar Structures to the E-To-H Helix Unit in the Globin-Like Fold Are Found in Other Helical Folds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rehmannia Glutinosa-Monocultured Rhizosphere Soil
Comparative Metaproteomic Analysis on Consecutively Rehmannia glutinosa-Monocultured Rhizosphere Soil Linkun Wu1,2, Haibin Wang1,2., Zhixing Zhang1,2., Rui Lin2,3, Zhongyi Zhang1,4, Wenxiong Lin1,2* 1 School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2 Agroecological Institute, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 3 College of Oceanography and Environmental Science, Xiamen University, Xiamen, Fujian, China, 4 Institute of Chinese Medicinal Materials, Henan Agriculture University, Zhengzhou, Henan, China Abstract Background: The consecutive monoculture for most of medicinal plants, such as Rehmannia glutinosa, results in a significant reduction in the yield and quality. There is an urgent need to study for the sustainable development of Chinese herbaceous medicine. Methodology/Principal Findings: Comparative metaproteomics of rhizosphere soil was developed and used to analyze the underlying mechanism of the consecutive monoculture problems of R. glutinosa. The 2D-gel patterns of protein spots for the soil samples showed a strong matrix dependency. Among the spots, 103 spots with high resolution and repeatability were randomly selected and successfully identified by MALDI TOF-TOF MS for a rhizosphere soil metaproteomic profile analysis. These proteins originating from plants and microorganisms play important roles in nutrient cycles and energy flow in rhizospheric soil ecosystem. They function in protein, nucleotide and secondary metabolisms, signal transduction and resistance. Comparative metaproteomics analysis revealed 33 differentially expressed protein spots in rhizosphere soil in response to increasing years of monoculture. Among them, plant proteins related to carbon and nitrogen metabolism and stress response, were mostly up-regulated except a down-regulated protein (glutathione S-transferase) involving detoxification. -
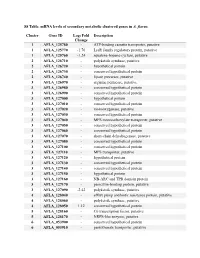
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

Electric Supporting Information (ESI) Crystal Structure and Functional
Electronic Supplementary Material (ESI) for Chemical Science. This journal is © The Royal Society of Chemistry 2018 Electric Supporting Information (ESI) Crystal structure and functional analysis of large-terpene synthase belonging to a newly found subclass Masahiro Fujihashi,a* Tsutomu Sato,b* Yuma Tanaka,a Daisuke Yamamoto,a Tomoyuki Nishi,b Daijiro Ueda,b Mizuki Murakami,b Yoko Yasuno,c Ai Sekihara,c Kazuma Fuku,c Tetsuro Shinadac & Kunio Mikia* a. Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: [email protected], [email protected] b. Department of Applied Biological Chemistry, Faculty of Agriculture, and Graduate School of Science and Technology, Niigata University, 8050 Ikarashi-2, Niigata 950-2181, Japan. E-mail: [email protected] c. Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi, Osaka 558-8585, Japan. S1 Table of Contents Experimental Procedures S3 General procedure S3 Vector construction, expression and purification of BalTS S3 Crystallization and crystallographic analysis of balTS S3 Oligomeric state analysis S4 Analysis of the dimer geometry S4 Vector construction, expression and purification of BsuTS S4 Homology modeling of BsuTS S4 Enzymatic assays S5 Isolation and identification of β-springen S5 Movie Captions S6 Author Contributions S6 Figures Fig. S1 S7 Fig. S2 S8 Fig. S3 S9 Fig. S4 S10 Fig. S5 S17 Fig. S6 S18 Fig. S7, S8 S19 Fig. S9 S20 Fig. S10 S21 Fig. S11 S22 Tables Table S1 S23 Table S2 S24 References S27 S2 Experimental Procedures General procedure NMR spectra were recorded using a Bruker DPX 400 spectrometer at 400 MHz for proton (1H) and 100 MHz for carbon (13C). -

Supporting Information High-Throughput Virtual Screening
Supporting Information High-Throughput Virtual Screening of Proteins using GRID Molecular Interaction Fields Simone Sciabola, Robert V. Stanton, James E. Mills, Maria M. Flocco, Massimo Baroni, Gabriele Cruciani, Francesca Perruccio and Jonathan S. Mason Contents Table S1 S2-S21 Figure S1 S22 * To whom correspondence should be addressed: Simone Sciabola, Pfizer Research Technology Center, Cambridge, 02139 MA, USA Phone: +1-617-551-3327; Fax: +1-617-551-3117; E-mail: [email protected] S1 Table S1. Description of the 990 proteins used as decoy for the Protein Virtual Screening analysis. PDB ID Protein family Molecule Res. (Å) 1n24 ISOMERASE (+)-BORNYL DIPHOSPHATE SYNTHASE 2.3 1g4h HYDROLASE 1,3,4,6-TETRACHLORO-1,4-CYCLOHEXADIENE HYDROLASE 1.8 1cel HYDROLASE(O-GLYCOSYL) 1,4-BETA-D-GLUCAN CELLOBIOHYDROLASE I 1.8 1vyf TRANSPORT PROTEIN 14 KDA FATTY ACID BINDING PROTEIN 1.85 1o9f PROTEIN-BINDING 14-3-3-LIKE PROTEIN C 2.7 1t1s OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.4 1t1r OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.3 1q0q OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 1.9 1jcy LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.9 1fww LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.85 1uk7 HYDROLASE 2-HYDROXY-6-OXO-7-METHYLOCTA-2,4-DIENOATE 1.7 1v11 OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.95 1x7w OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.73 1d0l TRANSFERASE 35KD SOLUBLE LYTIC TRANSGLYCOSYLASE 1.97 2bt4 LYASE 3-DEHYDROQUINATE DEHYDRATASE -
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

The Crystal Structure of Novel Chondroitin Lyase ODV-E66, A
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 587 (2013) 3943–3948 journal homepage: www.FEBSLetters.org The crystal structure of novel chondroitin lyase ODV-E66, a baculovirus envelope protein ⇑ Yoshirou Kawaguchi a, Nobuo Sugiura b, Koji Kimata c, Makoto Kimura a,d, Yoshimitsu Kakuta a,d, a Laboratory of Structural Biology, Graduate School of System Life Sciences, Kyushu University, 6-10-1 Hakozaki, Fukuoka 812-8581, Japan b Institute for Molecular Science of Medicine, Aichi Medical University, 1-1 Yazakokarimata, Nagakute, Aichi 480-1195, Japan c Research Complex for the Medicine Frontiers, Aichi Medical University, 1-1 Yazakokarimata, Nagakute, Aichi 480-1195, Japan d Faculty of Agriculture, Kyushu University, 6-10-1 Hakozaki, Fukuoka 812-8581, Japan article info abstract Article history: Chondroitin lyases have been known as pathogenic bacterial enzymes that degrade chondroitin. Received 5 August 2013 Recently, baculovirus envelope protein ODV-E66 was identified as the first reported viral chondroi- Revised 1 October 2013 tin lyase. ODV-E66 has low sequence identity with bacterial lyases at <12%, and unique characteris- Accepted 15 October 2013 tics reflecting the life cycle of baculovirus. To understand ODV-E66’s structural basis, the crystal Available online 26 October 2013 structure was determined and it was found that the structural fold resembled that of polysaccharide Edited by Christian Griesinger lyase 8 proteins and that the catalytic residues were also conserved. This structure enabled discus- sion of the unique substrate specificity and the stability of ODV-E66 as well as the host specificity of baculovirus. -

Hydroxylation of 1-Deoxypentalenic Acid Mediated by CYP105D7 (SAV 7469) of Streptomyces Avermitilis
The Journal of Antibiotics (2011) 64, 65–71 & 2011 Japan Antibiotics Research Association All rights reserved 0021-8820/11 $32.00 www.nature.com/ja ORIGINAL ARTICLE Pentalenic acid is a shunt metabolite in the biosynthesis of the pentalenolactone family of metabolites: hydroxylation of 1-deoxypentalenic acid mediated by CYP105D7 (SAV_7469) of Streptomyces avermitilis Satoshi Takamatsu1,4, Lian-Hua Xu2,4, Shinya Fushinobu2, Hirofumi Shoun2, Mamoru Komatsu1, David E Cane3 and Haruo Ikeda1 Pentalenic acid (1) has been isolated from many Streptomyces sp. as a co-metabolite of the sesquiterpenoid antibiotic pentalenolactone and related natural products. We have previously reported the identification of a 13.4-kb gene cluster in the genome of Streptomyces avermitilis implicated in the biosynthesis of the pentalenolactone family of metabolites consisting of 13 open reading frames. Detailed molecular genetic and biochemical studies have revealed that at least seven genes are involved in the biosynthesis of the newly discovered metabolites, neopentalenoketolactone, but no gene specifically dedicated to the formation of pentalenic acid (1) was evident in the same gene cluster. The wild-type strain of S. avermitilis, as well as its derivatives, mainly produce pentalenic acid (1), together with neopentalenoketolactone (9). Disruption of the sav7469 gene encoding a cytochrome P450 (CYP105D7), members of which class are associated with the hydroxylation of many structurally different compounds, abolished the production of pentalenic acid (1). The sav7469-deletion mutant derived from SUKA11 carrying pKU462Hptl-clusterDptlH accumulated 1-deoxypentalenic acid (5), but not pentalenic acid (1). Reintroduction of an extra-copy of the sav7469 gene to SUKA11 Dsav7469 carrying pKU462Hptl-clusterDptlH restored the production of pentalenic acid (1). -

Manual D'estil Per a Les Ciències De Laboratori Clínic
MANUAL D’ESTIL PER A LES CIÈNCIES DE LABORATORI CLÍNIC Segona edició Preparada per: XAVIER FUENTES I ARDERIU JAUME MIRÓ I BALAGUÉ JOAN NICOLAU I COSTA Barcelona, 14 d’octubre de 2011 1 Índex Pròleg Introducció 1 Criteris generals de redacció 1.1 Llenguatge no discriminatori per raó de sexe 1.2 Llenguatge no discriminatori per raó de titulació o d’àmbit professional 1.3 Llenguatge no discriminatori per raó d'ètnia 2 Criteris gramaticals 2.1 Criteris sintàctics 2.1.1 Les conjuncions 2.2 Criteris morfològics 2.2.1 Els articles 2.2.2 Els pronoms 2.2.3 Els noms comuns 2.2.4 Els noms propis 2.2.4.1 Els antropònims 2.2.4.2 Els noms de les espècies biològiques 2.2.4.3 Els topònims 2.2.4.4 Les marques registrades i els noms comercials 2.2.5 Els adjectius 2.2.6 El nombre 2.2.7 El gènere 2.2.8 Els verbs 2.2.8.1 Les formes perifràstiques 2.2.8.2 L’ús dels infinitius ser i ésser 2.2.8.3 Els verbs fer, realitzar i efectuar 2.2.8.4 Les formes i l’ús del gerundi 2.2.8.5 L'ús del verb haver 2.2.8.6 Els verbs haver i caldre 2.2.8.7 La forma es i se davant dels verbs 2.2.9 Els adverbis 2.2.10 Les locucions 2.2.11 Les preposicions 2.2.12 Els prefixos 2.2.13 Els sufixos 2.2.14 Els signes de puntuació i altres signes ortogràfics auxiliars 2.2.14.1 La coma 2.2.14.2 El punt i coma 2.2.14.3 El punt 2.2.14.4 Els dos punts 2.2.14.5 Els punts suspensius 2.2.14.6 El guionet 2.2.14.7 El guió 2.2.14.8 El punt i guió 2.2.14.9 L’apòstrof 2.2.14.10 L’interrogant 2 2.2.14.11 L’exclamació 2.2.14.12 Les cometes 2.2.14.13 Els parèntesis 2.2.14.14 Els claudàtors 2.2.14.15 -

Active Site of Chondroitin AC Lyase Revealed by the Structure Of
Biochemistry 2001, 40, 2359-2372 2359 Active Site of Chondroitin AC Lyase Revealed by the Structure of Enzyme-Oligosaccharide Complexes and Mutagenesis†,‡ Weijun Huang,§ Lorena Boju,§ Lydia Tkalec,|,⊥ Hongsheng Su,|,# Hyun-Ok Yang,3 Nur Sibel Gunay,3 Robert J. Linhardt,3 Yeong Shik Kim,O Allan Matte,§ and Miroslaw Cygler*,§ Biotechnology Research Institute, 6100 Royalmount AVenue, Montre´al, Que´bec H4P 2R2 Canada, Montreal Joint Centre for Structural Biology, Montre´al, Que´bec, Canada, IBEX Technologies Inc., 5485 Pare Street, Montre´al, Que´bec H4P 1P7 Canada, Department of Chemistry, DiVision of Medicinal Chemistry and Department of Chemical and Biochemical Engineering, The UniVersity of Iowa, 115 South Grand AVenue, PHAR S328, Iowa City, Iowa 52242-1112, and Natural Products Research Institute, Seoul National UniVersity, Seoul 110-460, Korea ReceiVed October 17, 2000; ReVised Manuscript ReceiVed December 18, 2000 ABSTRACT: The crystal structures of FlaVobacterium heparinium chondroitin AC lyase (chondroitinase AC; EC 4.2.2.5) bound to dermatan sulfate hexasaccharide (DShexa), tetrasaccharide (DStetra), and hyaluronic acid tetrasaccharide (HAtetra) have been refined at 2.0, 2.0, and 2.1 Å resolution, respectively. The structure of the Tyr234Phe mutant of AC lyase bound to a chondroitin sulfate tetrasaccharide (CStetra) has also been determined to 2.3 Å resolution. For each of these complexes, four (DShexa and CStetra) or two (DStetra and HAtetra) ordered sugars are visible in electron density maps. The lyase AC DShexa and CStetra complexes reveal binding at four subsites, -2, -1, +1, and +2, within a narrow and shallow protein channel. We suggest that subsites -2 and -1 together represent the substrate recognition area, +1 is the catalytic subsite and +1 and +2 together represent the product release area. -

Wo 2008/127291 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date PCT (10) International Publication Number 23 October 2008 (23.10.2008) WO 2008/127291 A2 (51) International Patent Classification: Jeffrey, J. [US/US]; 106 Glenview Drive, Los Alamos, GOlN 33/53 (2006.01) GOlN 33/68 (2006.01) NM 87544 (US). HARRIS, Michael, N. [US/US]; 295 GOlN 21/76 (2006.01) GOlN 23/223 (2006.01) Kilby Avenue, Los Alamos, NM 87544 (US). BURRELL, Anthony, K. [NZ/US]; 2431 Canyon Glen, Los Alamos, (21) International Application Number: NM 87544 (US). PCT/US2007/021888 (74) Agents: COTTRELL, Bruce, H. et al.; Los Alamos (22) International Filing Date: 10 October 2007 (10.10.2007) National Laboratory, LGTP, MS A187, Los Alamos, NM 87545 (US). (25) Filing Language: English (81) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of national protection available): AE, AG, AL, AM, AT,AU, AZ, BA, BB, BG, BH, BR, BW, BY,BZ, CA, CH, (30) Priority Data: CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, 60/850,594 10 October 2006 (10.10.2006) US ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, (71) Applicants (for all designated States except US): LOS LR, LS, LT, LU, LY,MA, MD, ME, MG, MK, MN, MW, ALAMOS NATIONAL SECURITY,LLC [US/US]; Los MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, Alamos National Laboratory, Lc/ip, Ms A187, Los Alamos, PT, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, SV, SY, NM 87545 (US). -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Supplementary Materials
Supplementary Materials OH 8 H H8 H C N OH 3 7 3 2 4 H1 1 5 O H4 O 6 H6’ H3 OH H6 H1’ H2 H5 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 ppm 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 ppm Figure S1. 1D 1H spectrum of 1-dglcnac and signal assignments ppm 2.0 H8 2.5 OH 8 H H C N OH 3 7 3 2 4 3.0 1 5 H1 O H5 O 6 H4 H3 OH 3.5 H6 H2 H6’ H1’ 4.0 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 2.0 ppm Figure S2. 1H-1H TOCSY 2D NMR spectra for 1-dglcnac and signal assignments ppm 3.0 H1 3.2 H5 H4 3.4 H3 3.6 H6 H6’ H2 3.8 H1’ 4.0 4.1 4.0 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 3.1 3.0 ppm Figure S3. 1H-1H COSY 2D NMR spectra for 1-dglcnac and signal assignment ppm -0.02 -0.01 0.00 0.01 0.02 H1’ H6’ H2 H6 H3 H4 H5 H1 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 3.1 ppm Figure S4. 1H-1H JRES 2D NMR spectra for 1-dglcnac and signal assignments ppm 20 C8(22.9) 40 OH 8 H H C N OH C2(53.2) 3 7 3 2 4 60 1 5 C6(63.2) C6(63.2) O 6 C1(69.2) C1(69.2) O C4(72.6) C3(77.0) OH 80 C5(82.7) 100 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 2.0 ppm Figure S5.