Characterization of Natural Product Biological Imprints for Computer-Aided Drug Design Applications Noe Sturm
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metabolic Engineering of Microbial Cell Factories for Biosynthesis of Flavonoids: a Review
molecules Review Metabolic Engineering of Microbial Cell Factories for Biosynthesis of Flavonoids: A Review Hanghang Lou 1,†, Lifei Hu 2,†, Hongyun Lu 1, Tianyu Wei 1 and Qihe Chen 1,* 1 Department of Food Science and Nutrition, Zhejiang University, Hangzhou 310058, China; [email protected] (H.L.); [email protected] (H.L.); [email protected] (T.W.) 2 Hubei Key Lab of Quality and Safety of Traditional Chinese Medicine & Health Food, Huangshi 435100, China; [email protected] * Correspondence: [email protected]; Tel.: +86-0571-8698-4316 † These authors are equally to this manuscript. Abstract: Flavonoids belong to a class of plant secondary metabolites that have a polyphenol structure. Flavonoids show extensive biological activity, such as antioxidative, anti-inflammatory, anti-mutagenic, anti-cancer, and antibacterial properties, so they are widely used in the food, phar- maceutical, and nutraceutical industries. However, traditional sources of flavonoids are no longer sufficient to meet current demands. In recent years, with the clarification of the biosynthetic pathway of flavonoids and the development of synthetic biology, it has become possible to use synthetic metabolic engineering methods with microorganisms as hosts to produce flavonoids. This article mainly reviews the biosynthetic pathways of flavonoids and the development of microbial expression systems for the production of flavonoids in order to provide a useful reference for further research on synthetic metabolic engineering of flavonoids. Meanwhile, the application of co-culture systems in the biosynthesis of flavonoids is emphasized in this review. Citation: Lou, H.; Hu, L.; Lu, H.; Wei, Keywords: flavonoids; metabolic engineering; co-culture system; biosynthesis; microbial cell factories T.; Chen, Q. -

Key Enzymes Involved in the Synthesis of Hops Phytochemical Compounds: from Structure, Functions to Applications
International Journal of Molecular Sciences Review Key Enzymes Involved in the Synthesis of Hops Phytochemical Compounds: From Structure, Functions to Applications Kai Hong , Limin Wang, Agbaka Johnpaul , Chenyan Lv * and Changwei Ma * College of Food Science and Nutritional Engineering, China Agricultural University, 17 Qinghua Donglu Road, Haidian District, Beijing 100083, China; [email protected] (K.H.); [email protected] (L.W.); [email protected] (A.J.) * Correspondence: [email protected] (C.L.); [email protected] (C.M.); Tel./Fax: +86-10-62737643 (C.M.) Abstract: Humulus lupulus L. is an essential source of aroma compounds, hop bitter acids, and xanthohumol derivatives mainly exploited as flavourings in beer brewing and with demonstrated potential for the treatment of certain diseases. To acquire a comprehensive understanding of the biosynthesis of these compounds, the primary enzymes involved in the three major pathways of hops’ phytochemical composition are herein critically summarized. Hops’ phytochemical components impart bitterness, aroma, and antioxidant activity to beers. The biosynthesis pathways have been extensively studied and enzymes play essential roles in the processes. Here, we introduced the enzymes involved in the biosynthesis of hop bitter acids, monoterpenes and xanthohumol deriva- tives, including the branched-chain aminotransferase (BCAT), branched-chain keto-acid dehydroge- nase (BCKDH), carboxyl CoA ligase (CCL), valerophenone synthase (VPS), prenyltransferase (PT), 1-deoxyxylulose-5-phosphate synthase (DXS), 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR), Geranyl diphosphate synthase (GPPS), monoterpene synthase enzymes (MTS), cinnamate Citation: Hong, K.; Wang, L.; 4-hydroxylase (C4H), chalcone synthase (CHS_H1), chalcone isomerase (CHI)-like proteins (CHIL), Johnpaul, A.; Lv, C.; Ma, C. -

Reduction by the Methylreductase System in Methanobacterium Bryantii WILLIAM B
JOURNAL OF BACTERIOLOGY, Jan. 1987, p. 87-92 Vol. 169, No. 1 0021-9193/87/010087-06$02.00/0 Copyright © 1987, American Society for Microbiology Inhibition by Corrins of the ATP-Dependent Activation and CO2 Reduction by the Methylreductase System in Methanobacterium bryantii WILLIAM B. WHITMAN'* AND RALPH S. WOLFE2 Department of Microbiology, University of Georgia, Athens, Georgia 30602,1 and Department of Microbiology, University ofIllinois, Urbana, Illinois 618012 Received 1 August 1986/Accepted 28 September 1986 Corrins inhibited the ATP-dependent activation of the methylreductase system and the methyl coenzyme M-dependent reduction of CO2 in extracts of Methanobacterium bryantii resolved from low-molecular-weight factors. The concentrations of cobinamides and cobamides required for one-half of maximal inhibition of the ATP-depen4ent activation were between 1 and 5 ,M. Cobinamides were more inhibitory at lower concentra- tiops than cobamides. Deoxyadenosylcobalamin was not inhibitory at concentrations up to 25 ,uM. The inhibition of CO2 reduction was competitive with respect to CO2. The concentration of methylcobalamin required for one-half of maximal inhibition was 5 ,M. Other cobamideg inhibited at similar concentrations, but diaquacobinami4e inhibited at lower concentrations. With respect to their affinities and specificities for corrins, inhibition of both the ATP-dependent activation'and CO2 reduction closely resembled the corrin- dependent activation of the methylreductase described in similar extracts (W. B. Whitman and R. S. Wolfe, J. Bacteriol. 164:165-172, 1985). However, whether the multiple effects of corrins are due to action at a single site is unknown. The effect of corrins (cobamides and cobinamides) on in CO2 reduction. -

Rehmannia Glutinosa-Monocultured Rhizosphere Soil
Comparative Metaproteomic Analysis on Consecutively Rehmannia glutinosa-Monocultured Rhizosphere Soil Linkun Wu1,2, Haibin Wang1,2., Zhixing Zhang1,2., Rui Lin2,3, Zhongyi Zhang1,4, Wenxiong Lin1,2* 1 School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2 Agroecological Institute, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 3 College of Oceanography and Environmental Science, Xiamen University, Xiamen, Fujian, China, 4 Institute of Chinese Medicinal Materials, Henan Agriculture University, Zhengzhou, Henan, China Abstract Background: The consecutive monoculture for most of medicinal plants, such as Rehmannia glutinosa, results in a significant reduction in the yield and quality. There is an urgent need to study for the sustainable development of Chinese herbaceous medicine. Methodology/Principal Findings: Comparative metaproteomics of rhizosphere soil was developed and used to analyze the underlying mechanism of the consecutive monoculture problems of R. glutinosa. The 2D-gel patterns of protein spots for the soil samples showed a strong matrix dependency. Among the spots, 103 spots with high resolution and repeatability were randomly selected and successfully identified by MALDI TOF-TOF MS for a rhizosphere soil metaproteomic profile analysis. These proteins originating from plants and microorganisms play important roles in nutrient cycles and energy flow in rhizospheric soil ecosystem. They function in protein, nucleotide and secondary metabolisms, signal transduction and resistance. Comparative metaproteomics analysis revealed 33 differentially expressed protein spots in rhizosphere soil in response to increasing years of monoculture. Among them, plant proteins related to carbon and nitrogen metabolism and stress response, were mostly up-regulated except a down-regulated protein (glutathione S-transferase) involving detoxification. -
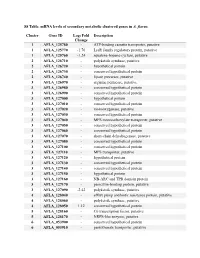
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

“Biosynthesis of Morphine in Mammals”
“Biosynthesis of Morphine in Mammals” D i s s e r t a t i o n zur Erlangung des akademischen Grades Doctor rerum naturalium (Dr. rer. nat.) vorgelegt der Naturwissenschaftlichen Fakultät I Biowissenschaften der Martin-Luther-Universität Halle-Wittenberg von Frau Nadja Grobe geb. am 21.08.1981 in Querfurt Gutachter /in 1. 2. 3. Halle (Saale), Table of Contents I INTRODUCTION ........................................................................................................1 II MATERIAL & METHODS ........................................................................................ 10 1 Animal Tissue ....................................................................................................... 10 2 Chemicals and Enzymes ....................................................................................... 10 3 Bacteria and Vectors ............................................................................................ 10 4 Instruments ........................................................................................................... 11 5 Synthesis ................................................................................................................ 12 5.1 Preparation of DOPAL from Epinephrine (according to DUNCAN 1975) ................. 12 5.2 Synthesis of (R)-Norlaudanosoline*HBr ................................................................. 12 5.3 Synthesis of [7D]-Salutaridinol and [7D]-epi-Salutaridinol ..................................... 13 6 Application Experiments ..................................................................................... -

1 Metabolic Dysfunction Is Restricted to the Sciatic Nerve in Experimental
Page 1 of 255 Diabetes Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy Oliver J. Freeman1,2, Richard D. Unwin2,3, Andrew W. Dowsey2,3, Paul Begley2,3, Sumia Ali1, Katherine A. Hollywood2,3, Nitin Rustogi2,3, Rasmus S. Petersen1, Warwick B. Dunn2,3†, Garth J.S. Cooper2,3,4,5* & Natalie J. Gardiner1* 1 Faculty of Life Sciences, University of Manchester, UK 2 Centre for Advanced Discovery and Experimental Therapeutics (CADET), Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Sciences Centre, Manchester, UK 3 Centre for Endocrinology and Diabetes, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, UK 4 School of Biological Sciences, University of Auckland, New Zealand 5 Department of Pharmacology, Medical Sciences Division, University of Oxford, UK † Present address: School of Biosciences, University of Birmingham, UK *Joint corresponding authors: Natalie J. Gardiner and Garth J.S. Cooper Email: [email protected]; [email protected] Address: University of Manchester, AV Hill Building, Oxford Road, Manchester, M13 9PT, United Kingdom Telephone: +44 161 275 5768; +44 161 701 0240 Word count: 4,490 Number of tables: 1, Number of figures: 6 Running title: Metabolic dysfunction in diabetic neuropathy 1 Diabetes Publish Ahead of Print, published online October 15, 2015 Diabetes Page 2 of 255 Abstract High glucose levels in the peripheral nervous system (PNS) have been implicated in the pathogenesis of diabetic neuropathy (DN). However our understanding of the molecular mechanisms which cause the marked distal pathology is incomplete. Here we performed a comprehensive, system-wide analysis of the PNS of a rodent model of DN. -

Mechanistic Implications for the Chorismatase Fkbo Based on the Crystal Structure
Mechanistic Implications for the Chorismatase FkbO Based on the Crystal Structure Puneet Juneja 1,†, Florian Hubrich 2,†, Kay Diederichs 1, Wolfram Welte 1 and Jennifer N. Andexer 2 1 - Department of Biology, University of Konstanz, D-78457 Konstanz, Germany 2 - Institute of Pharmaceutical Sciences, Albert-Ludwigs-University Freiburg, Albertstr 25, D-79104 Freiburg, Germany Correspondence to Jennifer N. Andexer: [email protected] http://dx.doi.org/10.1016/j.jmb.2013.09.006 Edited by M. Guss Abstract Chorismate-converting enzymes are involved in many biosynthetic pathways leading to natural products and can often be used as tools for the synthesis of chemical building blocks. Chorismatases such as FkbO from Streptomyces species catalyse the hydrolysis of chorismate yielding (dihydro)benzoic acid derivatives. In contrast to many other chorismate-converting enzymes, the structure and catalytic mechanism of a chorismatase had not been previously elucidated. Here we present the crystal structure of the chorismatase FkbO in complex with a competitive inhibitor at 1.08 Å resolution. FkbO is a monomer in solution and exhibits pseudo-3-fold symmetry; the structure of the individual domains indicates a possible connection to the trimeric RidA/YjgF family and related enzymes. The co-crystallised inhibitor led to the identification of FkbO's active site in the cleft between the central and the C-terminal domains. A mechanism for FkbO is proposed based on both interactions between the inhibitor and the surrounding amino acids and an FkbO structure with chorismate modelled in the active site. We suggest that the methylene group of the chorismate enol ether takes up a proton from an active-site glutamic acid residue, thereby initiating chorismate hydrolysis. -

Anti-Inflammatory Role of Curcumin in LPS Treated A549 Cells at Global Proteome Level and on Mycobacterial Infection
Anti-inflammatory Role of Curcumin in LPS Treated A549 cells at Global Proteome level and on Mycobacterial infection. Suchita Singh1,+, Rakesh Arya2,3,+, Rhishikesh R Bargaje1, Mrinal Kumar Das2,4, Subia Akram2, Hossain Md. Faruquee2,5, Rajendra Kumar Behera3, Ranjan Kumar Nanda2,*, Anurag Agrawal1 1Center of Excellence for Translational Research in Asthma and Lung Disease, CSIR- Institute of Genomics and Integrative Biology, New Delhi, 110025, India. 2Translational Health Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, 110067, India. 3School of Life Sciences, Sambalpur University, Jyoti Vihar, Sambalpur, Orissa, 768019, India. 4Department of Respiratory Sciences, #211, Maurice Shock Building, University of Leicester, LE1 9HN 5Department of Biotechnology and Genetic Engineering, Islamic University, Kushtia- 7003, Bangladesh. +Contributed equally for this work. S-1 70 G1 S 60 G2/M 50 40 30 % of cells 20 10 0 CURI LPSI LPSCUR Figure S1: Effect of curcumin and/or LPS treatment on A549 cell viability A549 cells were treated with curcumin (10 µM) and/or LPS or 1 µg/ml for the indicated times and after fixation were stained with propidium iodide and Annexin V-FITC. The DNA contents were determined by flow cytometry to calculate percentage of cells present in each phase of the cell cycle (G1, S and G2/M) using Flowing analysis software. S-2 Figure S2: Total proteins identified in all the three experiments and their distribution betwee curcumin and/or LPS treated conditions. The proteins showing differential expressions (log2 fold change≥2) in these experiments were presented in the venn diagram and certain number of proteins are common in all three experiments. -

Toxina PR En Penicillium Roqueforti
Universidad de León Instituto de Biotecnología de León Dpto. de Biología Molecular INBIOTEC Área de Microbiología TESIS DOCTORAL Caracterización Bioquímica y Molecular de la Biosíntesis del Antitumoral Andrastina y de la Toxina PR en Penicillium roqueforti . Pedro Iván Hidalgo Yanes León 2013 INFORME DEL DIRECTOR DE LA TESIS 1 (R.D. 99/2011, de 28 de enero y Normativa de la ULE) El Dr. D. Juan Francisco Martín Martín, el Dr. D. Ricardo Vicente Ullán y la Dra. Dña. Silvia Albillos García como Directores 2 de la Tesis Doctoral titulada “Caracterización Bioquímica y Molecular de la Biosíntesis del Antitumoral Andrastina y de la Toxina PR en Penicillium roqueforti ” realizada por D. Pedro Iván Hidalgo Yanes en el programa de doctorado Biología Molecular y Biotecnología, informan favorablemente el depósito de la misma, dado que reúne las condiciones necesarias para su defensa. León a ___ de__________ de _____________ D. Juan Francisco Martín Martín D. Ricardo Vicente Ullán Dña. Silvia Albillos García 1 Este impreso solamente se cumplimentará para los casos de tesis depositadas en papel. 2 Si la Tesis está dirigida por más de un Director tienen que constar los datos de cada uno y han de firmar todos ellos. ADMISIÓN A TRÁMITE DE LA TESIS DOCTORAL 3 (R.D. 99/2011, de 28 de enero y Normativa de la ULE) El órgano responsable del programa de doctorado _____________________________ ___________________________________________________________________________ en su reunión celebrada el día ___ de _____________ de ________ ha acordado dar su conformidad a la admisión a trámite de lectura de la Tesis Doctoral titulada “Caracterización Bioquímica y Molecular de la Biosíntesis del Antitumoral Andrastina y de la Toxina PR en Penicillium roqueforti ”, dirigida por el Dr. -

Transcriptome Analysis of Differential Gene Expression in Dichomitus Squalens
bioRxiv preprint doi: https://doi.org/10.1101/359646; this version posted June 30, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Transcriptome analysis of differential gene expression in Dichomitus squalens 2 during interspecific mycelial interactions and the potential link with laccase 3 induction 4 Zixuan Zhong1, Nannan Li1, Binghui He, Yasuo Igarashi, Feng Luo* 5 6 Research Center of Bioenergy and Bioremediation, College of Resources and Environment, 7 Southwest University, Beibei, Chongqing 400715, People’s Republic of China 8 9 *corresponding author: Feng Luo (FL) [email protected] 10 11 1These authors contributed equally to the paper 12 13 ABSTRACT 14 Interspecific mycelial interactions between white rot fungi are always accompanied by increased 15 production of laccase. In this study, the potential of white rot fungi Dichomitus squalens for 16 enhancing laccase production during interaction with two other white rot fungi Trametes 17 versicolor or Pleurotus ostreatus was identified. To probe the mechanism of laccase induction and 18 the role of laccase played during the combative interaction, we analyzed the laccase induction 19 response to stressful conditions during fungal interaction related to the differential gene expression 20 profile. We further confirmed the expression patterns of 16 selected genes by qRT-PCR analysis. 21 We noted that many differential expression genes (DEGs) encoding proteins were involved in 22 xenobiotics detoxification and ROS generation or reduction, including aldo/keto reductase, 23 glutathione S-transferases, cytochrome P450 enzymes, alcohol oxidases and dehydrogenase, 24 manganese peroxidase and laccase. -

Safrole and the Versatility of a Natural Biophore Lima, L
Artigo Safrole and the Versatility of a Natural Biophore Lima, L. M.* Rev. Virtual Quim., 2015, 7 (2), 495-538. Data de publicação na Web: 31 de dezembro de 2014 http://www.uff.br/rvq Safrol e a Versatilidade de um Biofóro Natural Resumo: Safrol (1) obtido de óleos essenciais de diferentes espécies vegetais tem ampla aplicação na indústria química como precursor sintético do butóxido de piperonila, piperonal e de fármacos como a tadalafila, cinoxacina e levodopa. Do ponto vista toxicológico, é considerado uma hepatotoxina por mecanismo de bioativação metabólica conduzindo a formação de intermediários eletrofílicos, e tem sido descrito como inibidor de diferentes isoenzimas da família CYP450. A versatilidade de sua estrutura, permitindo várias transformações químicas, e a natureza biofórica de sua unidade benzodioxola ou metilenodioxifenila conferem-lhe características singulares, que o tornam atrativo material de partida para a síntese de compostos com distintas atividades farmacológicas. Exemplos selecionados de compostos bioativos, naturais e sintéticos, contendo o sistema benzodioxola serão comentados, incluindo aqueles provenientes de contribuição específica do LASSBio®. Palavras-chave: Safrol; benzodioxola; metilenodioxi; CYP450; bióforo; compostos bioativos. Abstract Safrole (1), obtained from essential oils from different plant species, has wide application in the chemical industry as a synthetic precursor of piperonyl butoxide, piperonal and drugs such as tadalafil, cinoxacin and levodopa. From the toxicological point of view, it is considered a hepatotoxin through metabolic bioactivation, leading to the formation of electrophilic metabolites, and has been described as an inhibitor of different CYP450 isoenzymes. The versatility of its structure, allowing various chemical transformations, and the biophoric nature of its benzodioxole or methylenedioxy subunit, give it unique features and make it an attractive starting material for the synthesis of compounds with different pharmacological activities.