Diene Synthase for the Production of Novel Products
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Structure-Based Virtual Screening of Hypothetical Inhibitors of the Enzyme Longiborneol Synthase—A Potential Target to Reduce Fusarium Head Blight Disease
J Mol Model (2016) 22: 163 DOI 10.1007/s00894-016-3021-1 ORIGINAL PAPER Structure-based virtual screening of hypothetical inhibitors of the enzyme longiborneol synthase—a potential target to reduce Fusarium head blight disease E. Bresso1 & V. L ero ux 2 & M. Urban3 & K. E. Hammond-Kosack3 & B. Maigret2 & N. F. Martins1 Received: 17 December 2015 /Accepted: 27 May 2016 /Published online: 21 June 2016 # Springer-Verlag Berlin Heidelberg 2016 Abstract Fusarium head blight (FHB) is one of the most compounds from a library of 15,000 drug-like compounds. destructive diseases of wheat and other cereals worldwide. These putative inhibitors of longiborneol synthase provide a During infection, the Fusarium fungi produce mycotoxins that sound starting point for further studies involving molecular represent a high risk to human and animal health. Developing modeling coupled to biochemical experiments. This process small-molecule inhibitors to specifically reduce mycotoxin could eventually lead to the development of novel approaches levels would be highly beneficial since current treatments to reduce mycotoxin contamination in harvested grain. unspecifically target the Fusarium pathogen. Culmorin pos- sesses a well-known important synergistically virulence role Keywords Fusarium mycotoxins . Culmorin . Inhibitors . among mycotoxins, and longiborneol synthase appears to be a Homology modeling . Molecular dynamics . Ensemble key enzyme for its synthesis, thus making longiborneol syn- docking thase a particularly interesting target. This study aims to dis- cover potent and less toxic agrochemicals against FHB. These compounds would hamper culmorin synthesis by inhibiting Introduction longiborneol synthase. In order to select starting molecules for further investigation, we have conducted a structure- Fusarium head blight (FHB), caused by Fusarium based virtual screening investigation. -

Rehmannia Glutinosa-Monocultured Rhizosphere Soil
Comparative Metaproteomic Analysis on Consecutively Rehmannia glutinosa-Monocultured Rhizosphere Soil Linkun Wu1,2, Haibin Wang1,2., Zhixing Zhang1,2., Rui Lin2,3, Zhongyi Zhang1,4, Wenxiong Lin1,2* 1 School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2 Agroecological Institute, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 3 College of Oceanography and Environmental Science, Xiamen University, Xiamen, Fujian, China, 4 Institute of Chinese Medicinal Materials, Henan Agriculture University, Zhengzhou, Henan, China Abstract Background: The consecutive monoculture for most of medicinal plants, such as Rehmannia glutinosa, results in a significant reduction in the yield and quality. There is an urgent need to study for the sustainable development of Chinese herbaceous medicine. Methodology/Principal Findings: Comparative metaproteomics of rhizosphere soil was developed and used to analyze the underlying mechanism of the consecutive monoculture problems of R. glutinosa. The 2D-gel patterns of protein spots for the soil samples showed a strong matrix dependency. Among the spots, 103 spots with high resolution and repeatability were randomly selected and successfully identified by MALDI TOF-TOF MS for a rhizosphere soil metaproteomic profile analysis. These proteins originating from plants and microorganisms play important roles in nutrient cycles and energy flow in rhizospheric soil ecosystem. They function in protein, nucleotide and secondary metabolisms, signal transduction and resistance. Comparative metaproteomics analysis revealed 33 differentially expressed protein spots in rhizosphere soil in response to increasing years of monoculture. Among them, plant proteins related to carbon and nitrogen metabolism and stress response, were mostly up-regulated except a down-regulated protein (glutathione S-transferase) involving detoxification. -
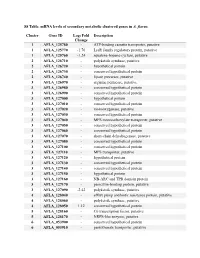
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

Characterization of a Cytochrome P450 Monooxygenase Gene Involved in the Biosynthesis of Geosmin in Penicillium Expansum
Open Archive TOULOUSE Archive Ouverte (OATAO) OATAO is an open access repository that collects the work of Toulouse researchers and makes it freely available over the web where possible. This is an author-deposited version published in : http://oatao.univ-toulouse.fr/ Eprints ID : 10812 To link to this article : DOI : 10.5897/AJMR11.1361 URL : http://academicjournals.org/journal/AJMR/article- abstract/841ECBA21771 To cite this version : Siddique, Muhammad Hussnain and Liboz, Thierry and Bacha, Nafees and Puel, Olivier and Mathieu, Florence and Lebrihi, Ahmed Characterization of a cytochrome P450 monooxygenase gene involved in the biosynthesis of geosmin in Penicillium expansum. (2012) African Journal of Microbiology Research, vol. 6 (n° 19). pp. 4122-4127. ISSN 1996-0808 Any correspondance concerning this service should be sent to the repository administrator: [email protected] Characterization of a cytochrome P450 monooxygenase gene involved in the biosynthesis of geosmin in Penicillium expansum Muhammad Hussnain Siddique1,2, Thierry Liboz1,2, Nafees Bacha3, Olivier Puel4, Florence Mathieu1,2 and Ahmed Lebrihi1,2,5* 1Université de Toulouse, INPT-UPS, Laboratoire de Génie Chimique, avenue de l’Agrobiopole, 31326 Castanet-Tolosan Cedex, France. 2Le Centre national de la recherche scientifique (CNRS), Laboratoire de Génie Chimique, 31030 Toulouse, France. 3Centre of Biotechnology and Microbiology, University of Peshawar, Pakistan. 4Institut National de la Recherche Agronomique (INRA), Laboratoire de Pharmacologie Toxicologie, 31931 Toulouse, France. 5Université Moulay Ismail, Marjane 2, BP 298, Meknes, Morocco. Geosmin is a terpenoid, an earthy-smelling substance associated with off-flavors in water and wine. The biosynthesis of geosmin is well characterized in bacteria, but little is known about its production in eukaryotes, especially in filamentous fungi. -

Product Profiles of Egyptian Henbane Premnaspirodiene
The Journal of Antibiotics (2016) 69, 524–533 & 2016 Japan Antibiotics Research Association All rights reserved 0021-8820/16 www.nature.com/ja ORIGINAL ARTICLE Biosynthetic potential of sesquiterpene synthases: product profiles of Egyptian Henbane premnaspirodiene synthase and related mutants Hyun Jo Koo1,3, Christopher R Vickery1,2,3,YiXu1, Gordon V Louie1, Paul E O'Maille1, Marianne Bowman1, Charisse M Nartey1, Michael D Burkart2 and Joseph P Noel1 The plant terpene synthase (TPS) family is responsible for the biosynthesis of a variety of terpenoid natural products possessing diverse biological functions. TPSs catalyze the ionization and, most commonly, rearrangement and cyclization of prenyl diphosphate substrates, forming linear and cyclic hydrocarbons. Moreover, a single TPS often produces several minor products in addition to a dominant product. We characterized the catalytic profiles of Hyoscyamus muticus premnaspirodiene synthase (HPS) and compared it with the profile of a closely related TPS, Nicotiana tabacum 5-epi-aristolochene synthase (TEAS). The profiles of two previously studied HPS and TEAS mutants, each containing nine interconverting mutations, dubbed HPS-M9 and TEAS- M9, were also characterized. All four TPSs were compared under varying temperature and pH conditions. In addition, we solved the X-ray crystal structures of TEAS and a TEAS quadruple mutant complexed with substrate and products to gain insight into the enzymatic features modulating product formation. These informative structures, along with product profiles, -

Searching for Novel Targets to Control Wheat Head Blight Disease—I-Protein Identification, 3D Modeling and Virtual Screening
Advances in Microbiology, 2016, 6, 811-830 http://www.scirp.org/journal/aim ISSN Online: 2165-3410 ISSN Print: 2165-3402 Searching for Novel Targets to Control Wheat Head Blight Disease—I-Protein Identification, 3D Modeling and Virtual Screening Natália F. Martins1, Emmanuel Bresso1, Roberto C. Togawa1, Martin Urban2, John Antoniw2, Bernard Maigret3, Kim Hammond-Kosack2 1EMBRAPA Recursos Genéticos e Biotecnologia Parque Estação Biológica, Brasília, Brazil 2Department of Plant Biology and Crop Science, Rothamsted Research, Harpenden, UK 3CNRS, LORIA, UMR 7503, Lorraine University, Nancy, France How to cite this paper: Martins, N.F., Abstract Bresso, E., Togawa, R.C., Urban, M., Anto- niw, J., Maigret, B. and Hammond-Kosack, Fusarium head blight (FHB) is a destructive disease of wheat and other cereals. FHB K. (2016) Searching for Novel Targets to occurs in Europe, North America and around the world causing significant losses in Control Wheat Head Blight Disease— production and endangers human and animal health. In this article, we provide the I-Protein Identification, 3D Modeling and strategic steps for the specific target selection for the phytopathogen system wheat- Virtual Screening. Advances in Microbiol- ogy, 6, 811-830. Fusarium graminearum. The economic impact of FHB leads to the need for innova- http://dx.doi.org/10.4236/aim.2016.611079 tion. Currently used fungicides have been shown to be effective over the years, but recently cereal infecting Fusaria have developed resistance. Our work presents a new Received: June 21, 2016 perspective on target selection to allow the development of new fungicides. We de- Accepted: September 11, 2016 veloped an innovative approach combining both genomic analysis and molecular Published: September 14, 2016 modeling to increase the discovery for new chemical compounds with both safety Copyright © 2016 by authors and and low environmental impact. -

Electric Supporting Information (ESI) Crystal Structure and Functional
Electronic Supplementary Material (ESI) for Chemical Science. This journal is © The Royal Society of Chemistry 2018 Electric Supporting Information (ESI) Crystal structure and functional analysis of large-terpene synthase belonging to a newly found subclass Masahiro Fujihashi,a* Tsutomu Sato,b* Yuma Tanaka,a Daisuke Yamamoto,a Tomoyuki Nishi,b Daijiro Ueda,b Mizuki Murakami,b Yoko Yasuno,c Ai Sekihara,c Kazuma Fuku,c Tetsuro Shinadac & Kunio Mikia* a. Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: [email protected], [email protected] b. Department of Applied Biological Chemistry, Faculty of Agriculture, and Graduate School of Science and Technology, Niigata University, 8050 Ikarashi-2, Niigata 950-2181, Japan. E-mail: [email protected] c. Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi, Osaka 558-8585, Japan. S1 Table of Contents Experimental Procedures S3 General procedure S3 Vector construction, expression and purification of BalTS S3 Crystallization and crystallographic analysis of balTS S3 Oligomeric state analysis S4 Analysis of the dimer geometry S4 Vector construction, expression and purification of BsuTS S4 Homology modeling of BsuTS S4 Enzymatic assays S5 Isolation and identification of β-springen S5 Movie Captions S6 Author Contributions S6 Figures Fig. S1 S7 Fig. S2 S8 Fig. S3 S9 Fig. S4 S10 Fig. S5 S17 Fig. S6 S18 Fig. S7, S8 S19 Fig. S9 S20 Fig. S10 S21 Fig. S11 S22 Tables Table S1 S23 Table S2 S24 References S27 S2 Experimental Procedures General procedure NMR spectra were recorded using a Bruker DPX 400 spectrometer at 400 MHz for proton (1H) and 100 MHz for carbon (13C). -

Origins of Isoprenoid Diversity: a Study of Structure-Function Relationships in Sesquiterpene Synthases" (2003)
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2003 ORIGINS OF ISOPRENOID DIVERSITY: A STUDY OF STRUCTURE- FUNCTION RELATIONSHIPS IN SESQUITERPENE SYNTHASES Bryan T. Greenhagen University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Greenhagen, Bryan T., "ORIGINS OF ISOPRENOID DIVERSITY: A STUDY OF STRUCTURE-FUNCTION RELATIONSHIPS IN SESQUITERPENE SYNTHASES" (2003). University of Kentucky Doctoral Dissertations. 440. https://uknowledge.uky.edu/gradschool_diss/440 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Bryan T. Greenhagen The Graduate School University of Kentucky 2003 1 ORIGINS OF ISOPRENOID DIVERSITY: A STUDY OF STRUCTURE-FUNCTION RELATIONSHIPS IN SESQUITERPENE SYNTHASES. ________________________________________ ABSTRACT OF DISSERTATION ________________________________________ A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Agriculture at the University of Kentucky by Bryan T. Greenhagen Lexington, KY Director: Dr. Joseph Chappell, Professor Lexington, KY Copyright © Bryan T. Greenhagen 2003 2 ABSTRACT OF DISSERTATION ORIGINS OF ISOPRENOID DIVERSITY: A STUDY OF STRUCTURE-FUNCTION RELATIONSHIPS IN SESQUITERPENE SYNTHASES. Plant sesquiterpene synthases catalyze the conversion of the linear substrate farnesyl diphosphate, FPP, into a remarkable array of secondary metabolites. These secondary metabolites in turn mediate a number of important interactions between plants and their environment, such as plant-plant, plant-insect and plant-pathogen interactions. -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000725805Cyc: Streptomyces xanthophaeus Cellular Overview Connections between pathways are omitted for legibility. -

1 AMINO ACIDS Commonly, 21 L-Amino Acids Encoded by DNA Represent the Building Blocks of Animal, Plant, and Microbial Proteins
1 AMINO ACIDS Commonly, 21 L-amino acids encoded by DNA represent the building blocks of animal, plant, and microbial proteins. The basic amino acids encountered in proteins are called proteinogenic amino acids 1.1). Biosynthesis of some of these amino acids proceeds by ribosomal processes only in microorganisms and plants and the ability to synthesize them is lacking in animals, including human beings. These amino acids have to be obtained in the diet (or produced by hydrolysis of body proteins) since they are required for normal good health and are referred to as essential amino acids. The essential amino acids are arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. The rest of encoded amino acids are referred to as non-essential amino acids (alanine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, proline, serine, and tyrosine). Arginine and histidine are classified as essential, sometimes as semi-essential amino acids, as their amount synthesized in the body is not sufficient for normal growth of children. Although it is itself non-essential, cysteine (classified as conditionally essential amino acid) can partly replace methionine, which is an essential amino acid. Similarly, tyrosine can partly replace phenylalanine. 1.1 The glutamic acid group 1.1.1 Glutamic acid and glutamine Free ammonium ions are toxic to living cells and are rapidly incorporated into organic compounds. One of such transformations is the reaction of ammonia with 2-oxoglutaric acid from the citric acid cycle to produce L-glutamic acid. This reaction is known as reductive amination. Glutamic acid is accordingly the amino acid generated first as both constituent of proteins and a biosynthetic precursor. -

Production of Plant-Associated Volatiles by Select Model and Industrially Important Streptomyces Spp
microorganisms Article Production of Plant-Associated Volatiles by Select Model and Industrially Important Streptomyces spp. 1, 2, 3 1 Zhenlong Cheng y, Sean McCann y, Nicoletta Faraone , Jody-Ann Clarke , E. Abbie Hudson 2, Kevin Cloonan 2, N. Kirk Hillier 2,* and Kapil Tahlan 1,* 1 Department of Biology, Memorial University of Newfoundland, St. John’s, NL A1B 3X9, Canada; [email protected] (Z.C.); [email protected] (J.-A.C.) 2 Department of Biology, Acadia University, Wolfville, NS B4P 2R6, Canada; [email protected] (S.M.); [email protected] (E.A.H.); [email protected] (K.C.) 3 Department of Chemistry, Acadia University, Wolfville, NS B4P 2R6, Canada; [email protected] * Correspondence: [email protected] (N.K.H.); [email protected] (K.T.) These authors contributed equally. y Received: 13 October 2020; Accepted: 9 November 2020; Published: 11 November 2020 Abstract: The Streptomyces produce a great diversity of specialized metabolites, including highly volatile compounds with potential biological activities. Volatile organic compounds (VOCs) produced by nine Streptomyces spp., some of which are of industrial importance, were collected and identified using gas chromatography–mass spectrometry (GC-MS). Biosynthetic gene clusters (BGCs) present in the genomes of the respective Streptomyces spp. were also predicted to match them with the VOCs detected. Overall, 33 specific VOCs were identified, of which the production of 16 has not been previously reported in the Streptomyces. Among chemical classes, the most abundant VOCs were terpenes, which is consistent with predicted biosynthetic capabilities. In addition, 27 of the identified VOCs were plant-associated, demonstrating that some Streptomyces spp. -

Supporting Information High-Throughput Virtual Screening
Supporting Information High-Throughput Virtual Screening of Proteins using GRID Molecular Interaction Fields Simone Sciabola, Robert V. Stanton, James E. Mills, Maria M. Flocco, Massimo Baroni, Gabriele Cruciani, Francesca Perruccio and Jonathan S. Mason Contents Table S1 S2-S21 Figure S1 S22 * To whom correspondence should be addressed: Simone Sciabola, Pfizer Research Technology Center, Cambridge, 02139 MA, USA Phone: +1-617-551-3327; Fax: +1-617-551-3117; E-mail: [email protected] S1 Table S1. Description of the 990 proteins used as decoy for the Protein Virtual Screening analysis. PDB ID Protein family Molecule Res. (Å) 1n24 ISOMERASE (+)-BORNYL DIPHOSPHATE SYNTHASE 2.3 1g4h HYDROLASE 1,3,4,6-TETRACHLORO-1,4-CYCLOHEXADIENE HYDROLASE 1.8 1cel HYDROLASE(O-GLYCOSYL) 1,4-BETA-D-GLUCAN CELLOBIOHYDROLASE I 1.8 1vyf TRANSPORT PROTEIN 14 KDA FATTY ACID BINDING PROTEIN 1.85 1o9f PROTEIN-BINDING 14-3-3-LIKE PROTEIN C 2.7 1t1s OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.4 1t1r OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.3 1q0q OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 1.9 1jcy LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.9 1fww LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.85 1uk7 HYDROLASE 2-HYDROXY-6-OXO-7-METHYLOCTA-2,4-DIENOATE 1.7 1v11 OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.95 1x7w OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.73 1d0l TRANSFERASE 35KD SOLUBLE LYTIC TRANSGLYCOSYLASE 1.97 2bt4 LYASE 3-DEHYDROQUINATE DEHYDRATASE