Origins of Isoprenoid Diversity: a Study of Structure-Function Relationships in Sesquiterpene Synthases" (2003)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 AMINO ACIDS Commonly, 21 L-Amino Acids Encoded by DNA Represent the Building Blocks of Animal, Plant, and Microbial Proteins
1 AMINO ACIDS Commonly, 21 L-amino acids encoded by DNA represent the building blocks of animal, plant, and microbial proteins. The basic amino acids encountered in proteins are called proteinogenic amino acids 1.1). Biosynthesis of some of these amino acids proceeds by ribosomal processes only in microorganisms and plants and the ability to synthesize them is lacking in animals, including human beings. These amino acids have to be obtained in the diet (or produced by hydrolysis of body proteins) since they are required for normal good health and are referred to as essential amino acids. The essential amino acids are arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. The rest of encoded amino acids are referred to as non-essential amino acids (alanine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, proline, serine, and tyrosine). Arginine and histidine are classified as essential, sometimes as semi-essential amino acids, as their amount synthesized in the body is not sufficient for normal growth of children. Although it is itself non-essential, cysteine (classified as conditionally essential amino acid) can partly replace methionine, which is an essential amino acid. Similarly, tyrosine can partly replace phenylalanine. 1.1 The glutamic acid group 1.1.1 Glutamic acid and glutamine Free ammonium ions are toxic to living cells and are rapidly incorporated into organic compounds. One of such transformations is the reaction of ammonia with 2-oxoglutaric acid from the citric acid cycle to produce L-glutamic acid. This reaction is known as reductive amination. Glutamic acid is accordingly the amino acid generated first as both constituent of proteins and a biosynthetic precursor. -

Diene Synthase for the Production of Novel Products
Engineering Selina-4(15),7(11)-diene synthase for the Production of Novel Products Emily Turri Submitted in accordance with the requirements for the degree of Doctor of Philosophy The University of Leeds Astbury Centre for Structural Molecular Biology September 2020 The candidate confirms that the work submitted is her own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. The right of Emily Turri to be identified as Author of this work has been asserted by her in accordance with the Copyright, Designs and Patents Act 1988. © 2020 The University of Leeds and Emily Turri 2 Acknowledgements Throughout my PhD I have been extremely lucky to receive support and encouragement from family, friends, colleagues and the university. Without these people and their support, this thesis would not be possible. First, I would like to thank my supervisors Alan Berry and Adam Nelson for their encouragement, guidance and patience over these four years. I would also like to thank Glyn Hemsworth and Nasir Khan for their continued advice and support. I am very grateful to Glyn Hemsworth, Sheena Radford, David Brockwell and John Blacker for letting me use their equipment while offering me direction to give me the best results. In addition, I would like to thank James Ault, Rachel George, Mary Bayana, Mark Howard and Ricardo Labes for their technical assistance and biochemical analysis. My PhD experience would have been nothing without the present and past members of the Hemrry’s and Radford’s with specific thanks to Jess, Alex, Jess and Dan. -

David E. Cane Publications 1. EJ Corey and David E. Cane
David E. Cane Publications 1. E. J. Corey and David E. Cane, "The Synthesis of Olefins from β-Hydroxyphosphonamides. Stereochemistry and Extension to the Formation of Conjugated Dienes," J. Org. Chem., 34, 3053 (1969). 2. E. J. Corey and David E. Cane, "The Metalation-Alkylation of Ynamines," J. Org.Chem., 35, 3405 (1970). 3. E. J. Corey and David E. Cane, "A New Method for the Controlled Hydroxymethylation of Ketones," J. Org. Chem., 36, 3070 (1971). 4. E. J. Corey, David E. Cane, and Lawrence Libit, "The Synthesis of Racemic α-trans- and β-trans-Bergamotene," J. Am. Chem. Soc., 93, 7013, (1971). 5. D. Arigoni, David E. Cane, Beat Mueller and Christopher Tamm, "The Mode of Incorporation of Farnesyl Pyrophosphate into Verrucarol," Helv. Chim. Acta, 56, 2946 (1973). 6. David E. Cane and Ronald H. Levin, "Application of Carbon-13 Magnetic Resonance to Isoprenoid Biosynthesis. I. Ovalicin," J. Am. Chem. Soc., 97, 1982 (1975). 7. David E. Cane and Ronald H. Levin, "Application of Carbon-13 Magnetic Resonance to Isoprenoid Biosynthesis. II. Ovalicin and the Use of Double-Labeled Mevalonate," J. Am. Chem. Soc., 98, 1183 (1976). 8. David E. Cane and Robert B. Nachbar, "Biosynthesis of Fomannosin from [1,2-13C]-Acetate," Tetrahedron Lett. 2097-2100 (1976). 9. David E. Cane and G. G. S. King, "The Biosynthesis of Ovalicin: Isolation of β-trans-Bergamotene," Tetrahedron Lett.., 4737 (1976). 10. David E. Cane and Stephen L. Buchwald, "Application of Deuterium Magnetic Resonance to Biosynthetic Studies I; Biosynthesis of Ovalicin," J. Am. Chem. Soc., 99, 6132 (1977). 11. David E. -

Phytohormones and Volatile Organic Compounds, Like Geosmin, in the Ectomycorrhiza of Tricholoma Vaccinum and Norway Spruce (Picea Abies)
Mycorrhiza (2021) 31:173–188 https://doi.org/10.1007/s00572-020-01005-2 ORIGINAL ARTICLE Phytohormones and volatile organic compounds, like geosmin, in the ectomycorrhiza of Tricholoma vaccinum and Norway spruce (Picea abies) Oluwatosin Abdulsalam1 · Katharina Wagner1 · Sophia Wirth1 · Maritta Kunert2 · Anja David2 · Mario Kallenbach2 · Wilhelm Boland2 · Erika Kothe1 · Katrin Krause1 Received: 21 June 2020 / Accepted: 11 November 2020 / Published online: 18 November 2020 © The Author(s) 2020 Abstract The ectomycorrhizospheric habitat contains a diverse pool of organisms, including the host plant, mycorrhizal fungi, and other rhizospheric microorganisms. Diferent signaling molecules may infuence the ectomycorrhizal symbiosis. Here, we investigated the potential of the basidiomycete Tricholoma vaccinum to produce communication molecules for the interac- tion with its coniferous host, Norway spruce (Picea abies). We focused on the production of volatile organic compounds and phytohormones in axenic T. vaccinum cultures, identifed the potential biosynthesis genes, and investigated their expression by RNA-Seq analyses. T. vaccinum released volatiles not usually associated with fungi, like limonene and β-barbatene, and geosmin. Using stable isotope labeling, the biosynthesis of geosmin was elucidated. The geosmin biosynthesis gene ges1 of T. vaccinum was identifed, and up-regulation was scored during mycorrhiza, while a diferent regulation was seen with mycorrhizosphere bacteria. The fungus also released the volatile phytohormone ethylene and excreted salicylic and abscisic acid as well as jasmonates into the medium. The tree excreted the auxin, indole-3-acetic acid, and its biosynthesis intermediate, indole-3-acetamide, as well as salicylic acid with its root exudates. These compounds could be shown for the frst time in exudates as well as in soil of a natural ectomycorrhizospheric habitat. -

Structural and Functional Studies of Two Bacterial Terpene Cyclases: Geosmin Synthase and Epi-Isozizaene Synthase
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Structural And Functional Studies Of Two Bacterial Terpene Cyclases: Geosmin Synthase And Epi-Isozizaene Synthase Golda Harris Barrow University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, and the Chemistry Commons Recommended Citation Barrow, Golda Harris, "Structural And Functional Studies Of Two Bacterial Terpene Cyclases: Geosmin Synthase And Epi-Isozizaene Synthase" (2017). Publicly Accessible Penn Dissertations. 2181. https://repository.upenn.edu/edissertations/2181 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2181 For more information, please contact [email protected]. Structural And Functional Studies Of Two Bacterial Terpene Cyclases: Geosmin Synthase And Epi-Isozizaene Synthase Abstract Terpene cyclases convert acyclic isoprenoid precursors into complex cyclic terpenoid compounds. Hydrophobic active site contours in terpene cyclases direct cyclization reactions through a cascade of carbocation intermediates by serving as templates for terpenoid product structure. Both local molecular structure at the active site and global protein structure encompassing domain organization and oligomerization state have effects on catalytic function in terpene cyclases. The goal of this work is characterization of the effects of local structural changes to the active site of a terpene cyclase, and a thorough understanding of the structure-function relationship of active site structure and domain organization in two terpene cyclases, geosmin synthase (ScGS) and epi-isozizaene synthase (EIZS) from Streptomyces coelicolor. Geosmin synthase (ScGS) is a bifunctional class I sesquiterpene cyclase that catalyzes the conversion of FPP to germacradienol, germacrene D, and geosmin in unique cyclization and cyclization-fragmentation reactions occurring in separate active sites. -

The Enzyme Database: New Enzymes 06/27/2006 05:11 PM
The Enzyme Database: New Enzymes 06/27/2006 05:11 PM Home Search Enzymes by Class New/Amended Enzymes Statistics Forms Advanced Search Information Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) Proposed Changes to the Enzyme List The entries below are proposed additions and amendments to the Enzyme Nomenclature list. They were prepared for the NC- IUBMB by Keith Tipton, Sinéad Boyce, Gerry Moss and Hal Dixon, with occasional help from other Committee members, and were put on the web by Gerry Moss. Comments and suggestions on these draft entries should be sent to Professor K.F. Tipton and Dr S. Boyce (Department of Biochemistry, Trinity College Dublin, Dublin 2, Ireland) by 20 May 2006, after which, the entries will be made official and will be incorporated into the main enzyme list. To prevent confusion please do not quote new EC numbers until they are incorporated into the main list. Many thanks to those of you who have submitted details of new enzymes or updates to existing enzymes. An asterisk before 'EC' indicates that this is an amendment to an existing enzyme rather than a new enzyme entry. Contents *EC 1.1.1.262 4-hydroxythreonine-4-phosphate dehydrogenase EC 1.1.1.289 sorbose reductase EC 1.1.1.290 4-phosphoerythronate dehydogenase EC 1.1.99.19 transferred *EC 1.2.1.10 acetaldehyde dehydrogenase (acetylating) EC 1.2.1.71 succinylglutamate-semialdehyde dehydrogenase EC 1.2.1.72 erythrose-4-phosphate dehydrogenase EC 1.2.99.1 transferred *EC 1.3.99.19 quinoline-4-carboxylate 2-oxidoreductase -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

Springer Handbook of Enzymes
Dietmar Schomburg Ida Schomburg (Eds.) Springer Handbook of Enzymes Alphabetical Name Index 1 23 © Springer-Verlag Berlin Heidelberg New York 2010 This work is subject to copyright. All rights reserved, whether in whole or part of the material con- cerned, specifically the right of translation, printing and reprinting, reproduction and storage in data- bases. The publisher cannot assume any legal responsibility for given data. Commercial distribution is only permitted with the publishers written consent. Springer Handbook of Enzymes, Vols. 1–39 + Supplements 1–7, Name Index 2.4.1.60 abequosyltransferase, Vol. 31, p. 468 2.7.1.157 N-acetylgalactosamine kinase, Vol. S2, p. 268 4.2.3.18 abietadiene synthase, Vol. S7,p.276 3.1.6.12 N-acetylgalactosamine-4-sulfatase, Vol. 11, p. 300 1.14.13.93 (+)-abscisic acid 8’-hydroxylase, Vol. S1, p. 602 3.1.6.4 N-acetylgalactosamine-6-sulfatase, Vol. 11, p. 267 1.2.3.14 abscisic-aldehyde oxidase, Vol. S1, p. 176 3.2.1.49 a-N-acetylgalactosaminidase, Vol. 13,p.10 1.2.1.10 acetaldehyde dehydrogenase (acetylating), Vol. 20, 3.2.1.53 b-N-acetylgalactosaminidase, Vol. 13,p.91 p. 115 2.4.99.3 a-N-acetylgalactosaminide a-2,6-sialyltransferase, 3.5.1.63 4-acetamidobutyrate deacetylase, Vol. 14,p.528 Vol. 33,p.335 3.5.1.51 4-acetamidobutyryl-CoA deacetylase, Vol. 14, 2.4.1.147 acetylgalactosaminyl-O-glycosyl-glycoprotein b- p. 482 1,3-N-acetylglucosaminyltransferase, Vol. 32, 3.5.1.29 2-(acetamidomethylene)succinate hydrolase, p. 287 Vol. -

Nature As Organic Chemist
The Journal of Antibiotics (2016) 69, 473–485 & 2016 Japan Antibiotics Research Association All rights reserved 0021-8820/16 www.nature.com/ja EDITORIAL Nature as organic chemist The Journal of Antibiotics (2016) 69, 473–485; doi:10.1038/ja.2016.55 y the time I entered Harvard College in the fall of 1962, I knew, studying the biological conversion of squalene oxide to lanosterol, Bor thought I knew, that I wanted to become a scientist. In fact, I was at that time more interested in the development of new synthetic I had very little idea what that actually meant, nor did I even know methods. Over the next few years, I carried out research on a number what branch of science to choose. My interest in chemistry was quickly of novel organolithium reagents, and eventually took up the synthesis kindled, however, by Prof Leonard K Nash’s introductory course in of the bicyclic sesquiterpene β-trans-bergamotene (1), the structure of general chemistry. I was not only smitten by the subject itself but also which had been assigned to a constituent of valerian root oil I was inspired by Prof Nash’s informative and entertaining lectures, (Figure 1).1 I finally completed my synthesis late one night and his warm and scintillating personality. Indeed for many years in February 1971. At 3:00 AM that morning I ran down to the thereafter, Prof Nash continued to be a source of encouragement and NMR room to record my triumph, only to be stunned when the advice on both science and the art of teaching. -
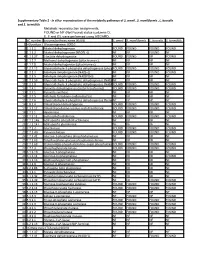
Supplementary Table 2 - in Silico Reconstruction of the Metabolic Pathways of S
Supplementary Table 2 - In silico reconstruction of the metabolic pathways of S. amnii , S. moniliformis , L. buccalis and S. termiditis Metabolic reconstruction assignments, FOUND or NF (Not Found) status (columns D, E, F and G), were performed using ASGARD, EC number Enzyme/pathway name (KEGG) S. amnii S. moniliformis L. buccalis S. termiditis 1 >Glycolysis / Gluconeogenesis 00010 2 1.1.1.1 Alcohol dehydrogenase. FOUND FOUND FOUND FOUND 3 1.1.1.2 Alcohol dehydrogenase (NADP(+)). NF NF FOUND NF 4 1.1.1.27 L-lactate dehydrogenase. FOUND FOUND NF FOUND 5 1.1.2.7 Methanol dehydrogenase (cytochrome c). NF NF NF NF 6 1.1.2.8 Alcohol dehydrogenase (cytochrome c). NF NF NF NF 7 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating).FOUND FOUND FOUND FOUND 8 1.2.1.3 Aldehyde dehydrogenase (NAD(+)). NF NF FOUND FOUND 9 1.2.1.5 Aldehyde dehydrogenase (NAD(P)(+)). NF NF NF NF 10 1.2.1.59 Glyceraldehyde-3-phosphate dehydrogenase (NAD(P)(+))NF (phosphorylating).NF NF NF 11 1.2.1.9 Glyceraldehyde-3-phosphate dehydrogenase (NADP(+)).FOUND FOUND FOUND FOUND 12 1.2.4.1 Pyruvate dehydrogenase (acetyl-transferring). FOUND FOUND FOUND FOUND 13 1.2.7.1 Pyruvate synthase. NF NF NF NF 14 1.2.7.5 Aldehyde ferredoxin oxidoreductase. NF NF NF NF 15 1.2.7.6 Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin).NF NF NF NF 16 1.8.1.4 Dihydrolipoyl dehydrogenase. FOUND FOUND FOUND FOUND 17 2.3.1.12 Dihydrolipoyllysine-residue acetyltransferase. FOUND FOUND FOUND FOUND 18 2.7.1.1 Hexokinase. -

WO 2017/139420 Al 17 August 2017 (17.08.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/139420 Al 17 August 2017 (17.08.2017) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C07C 45/27 (2006.01) C12P 19/24 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KH, KN, PCT/US20 17/0 17069 KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, (22) International Filing Date: MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, 8 February 2017 (08.02.2017) NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, (25) Filing Language: English TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, (26) Publication Language: English ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 62/292,924 9 February 2016 (09.02.2016) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (71) Applicant: KEMBIOTIX LLC [US/US]; 325 Speen TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, Street #404, Natick, MA 01760 (US). -

Biosynthetic Engineering of Plantazolicin Natural Products
BIOSYNTHETIC ENGINEERING OF PLANTAZOLICIN NATURAL PRODUCTS BY CAITLIN D. DEANE DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2016 Urbana, Illinois Doctoral Committee: Associate Professor Douglas A. Mitchell, Chair Professor Wilfred A. van der Donk Professor William W. Metcalf Professor Huimin Zhao ABSTRACT Plantazolicin (PZN) is a ribosomally synthesized and post-translationally modified peptide (RiPP) natural product that exhibits extraordinarily narrow-spectrum antibacterial activity towards the causative agent of anthrax, Bacillus anthracis. During PZN biosynthesis, a cyclodehydratase catalyzes cyclization of cysteine, serine, and threonine residues in the PZN precursor peptide (BamA) to azolines. Subsequently, a dehydrogenase then oxidizes most of these azolines to thiazoles and (methyl)oxazoles. The final biosynthetic steps consist of leader peptide removal and dimethylation of the nascent N-terminus. To simultaneously establish the structure−activity relationship of PZN and the substrate tolerance of the biosynthetic pathway, an Escherichia coli expression strain was engineered to heterologously produce PZN analogs. 72 variant BamA peptides were screened by mass spectrometry to assess post-translational modification and export by E. coli, from which 29 PZN variants were detected. The modifying enzymes were exquisitely selective, installing heterocycles only at predefined positions within