Nature As Organic Chemist
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biomolecules
CHAPTER 3 Biomolecules 3.1 Carbohydrates In the previous chapter you have learnt about the cell and 3.2 Fatty Acids and its organelles. Each organelle has distinct structure and Lipids therefore performs different function. For example, cell membrane is made up of lipids and proteins. Cell wall is 3.3 Amino Acids made up of carbohydrates. Chromosomes are made up of 3.4 Protein Structure protein and nucleic acid, i.e., DNA and ribosomes are made 3.5 Nucleic Acids up of protein and nucleic acids, i.e., RNA. These ingredients of cellular organelles are also called macromolecules or biomolecules. There are four major types of biomolecules— carbohydrates, proteins, lipids and nucleic acids. Apart from being structural entities of the cell, these biomolecules play important functions in cellular processes. In this chapter you will study the structure and functions of these biomolecules. 3.1 CARBOHYDRATES Carbohydrates are one of the most abundant classes of biomolecules in nature and found widely distributed in all life forms. Chemically, they are aldehyde and ketone derivatives of the polyhydric alcohols. Major role of carbohydrates in living organisms is to function as a primary source of energy. These molecules also serve as energy stores, 2021-22 Chapter 3 Carbohydrade Final 30.018.2018.indd 50 11/14/2019 10:11:16 AM 51 BIOMOLECULES metabolic intermediates, and one of the major components of bacterial and plant cell wall. Also, these are part of DNA and RNA, which you will study later in this chapter. The cell walls of bacteria and plants are made up of polymers of carbohydrates. -

Structure-Based Virtual Screening of Hypothetical Inhibitors of the Enzyme Longiborneol Synthase—A Potential Target to Reduce Fusarium Head Blight Disease
J Mol Model (2016) 22: 163 DOI 10.1007/s00894-016-3021-1 ORIGINAL PAPER Structure-based virtual screening of hypothetical inhibitors of the enzyme longiborneol synthase—a potential target to reduce Fusarium head blight disease E. Bresso1 & V. L ero ux 2 & M. Urban3 & K. E. Hammond-Kosack3 & B. Maigret2 & N. F. Martins1 Received: 17 December 2015 /Accepted: 27 May 2016 /Published online: 21 June 2016 # Springer-Verlag Berlin Heidelberg 2016 Abstract Fusarium head blight (FHB) is one of the most compounds from a library of 15,000 drug-like compounds. destructive diseases of wheat and other cereals worldwide. These putative inhibitors of longiborneol synthase provide a During infection, the Fusarium fungi produce mycotoxins that sound starting point for further studies involving molecular represent a high risk to human and animal health. Developing modeling coupled to biochemical experiments. This process small-molecule inhibitors to specifically reduce mycotoxin could eventually lead to the development of novel approaches levels would be highly beneficial since current treatments to reduce mycotoxin contamination in harvested grain. unspecifically target the Fusarium pathogen. Culmorin pos- sesses a well-known important synergistically virulence role Keywords Fusarium mycotoxins . Culmorin . Inhibitors . among mycotoxins, and longiborneol synthase appears to be a Homology modeling . Molecular dynamics . Ensemble key enzyme for its synthesis, thus making longiborneol syn- docking thase a particularly interesting target. This study aims to dis- cover potent and less toxic agrochemicals against FHB. These compounds would hamper culmorin synthesis by inhibiting Introduction longiborneol synthase. In order to select starting molecules for further investigation, we have conducted a structure- Fusarium head blight (FHB), caused by Fusarium based virtual screening investigation. -

Sugars As the Source of Energized Carbon for Abiogenesis
Astrobiology Science Conference 2010 (2010) 5095.pdf SUGARS AS THE SOURCE OF ENERGIZED CARBON FOR ABIOGENESIS. A. L. Weber, SETI Institute, NASA Ames Research Center, Mail Stop 239-4, Moffett Field, CA, 94035-1000, [email protected] Abstract: As shown in Figure 1, abiogenesis has sev- eral requirements: (A) a source of organic substrates and chemical energy that drives the synthesis of (B) useful small molecules (ammonia, monomers, metabo- lites, energy molecules), and (C) a second synthetic processs that yields large replicating and catalytic polymers that control (D) the growth and maintenance of a primitive protocell. Furthermore, the required chemical energy must be sustained and effectively coupled to individual reactions to drive biosynthesis at a rate that counters chemical degradation. Energy coupling would have been especially difficult during the origin of life before the development of powerful enzyme catalysts with 3-D active sites. To solve this energy coupling problem we have investigated abio- genesis using sugar substrates whose energized carbon groups drive spontaneous synthetic self-transformation reactions that yield: biometabolites, catalytic mole- cules, energy-rich thioesters, amino acids, plausible alternative nucleobases and cell-like microstructures [1-8]. Recently, we demonstrated that sugars drive the synthesis of ammonia from nitrite [9]. The ability of sugars to drive ammonia synthesis provides a way to generate ammonia at microscopic sites of sugar-based origins processes, thereby eliminating the need for a planet-wide source of photochemically unstable am- monia. Figure 1. Major Synthetic Processes of Abiogenesis. [1] Weber A. L. (1998) Orig. Life Evol. Biosph., 28, 259-270. [2] Weber A. -

Rehmannia Glutinosa-Monocultured Rhizosphere Soil
Comparative Metaproteomic Analysis on Consecutively Rehmannia glutinosa-Monocultured Rhizosphere Soil Linkun Wu1,2, Haibin Wang1,2., Zhixing Zhang1,2., Rui Lin2,3, Zhongyi Zhang1,4, Wenxiong Lin1,2* 1 School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2 Agroecological Institute, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 3 College of Oceanography and Environmental Science, Xiamen University, Xiamen, Fujian, China, 4 Institute of Chinese Medicinal Materials, Henan Agriculture University, Zhengzhou, Henan, China Abstract Background: The consecutive monoculture for most of medicinal plants, such as Rehmannia glutinosa, results in a significant reduction in the yield and quality. There is an urgent need to study for the sustainable development of Chinese herbaceous medicine. Methodology/Principal Findings: Comparative metaproteomics of rhizosphere soil was developed and used to analyze the underlying mechanism of the consecutive monoculture problems of R. glutinosa. The 2D-gel patterns of protein spots for the soil samples showed a strong matrix dependency. Among the spots, 103 spots with high resolution and repeatability were randomly selected and successfully identified by MALDI TOF-TOF MS for a rhizosphere soil metaproteomic profile analysis. These proteins originating from plants and microorganisms play important roles in nutrient cycles and energy flow in rhizospheric soil ecosystem. They function in protein, nucleotide and secondary metabolisms, signal transduction and resistance. Comparative metaproteomics analysis revealed 33 differentially expressed protein spots in rhizosphere soil in response to increasing years of monoculture. Among them, plant proteins related to carbon and nitrogen metabolism and stress response, were mostly up-regulated except a down-regulated protein (glutathione S-transferase) involving detoxification. -
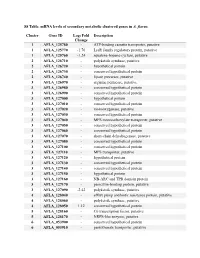
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

An Overview of Biosynthesis Pathways – Inspiration for Pharmaceutical and Agrochemical Discovery
An Overview of Biosynthesis Pathways – Inspiration for Pharmaceutical and Agrochemical Discovery Alan C. Spivey [email protected] 19th Oct 2019 Lessons in Synthesis - Azadirachtin • Azadirachtin is a potent insect anti-feedant from the Indian neem tree: – exact biogenesis unknown but certainly via steroid modification: O MeO C OAc O 2 H O OH O H O OH 12 O O C 11 O 14 OH oxidative 8 O H 7 cleavage highly hindered C-C bond HO OH AcO OH AcO OH for synthesis! H H of C ring H MeO2C O AcO H tirucallol azadirachtanin A azadirachtin (cf. lanosterol) (a limanoid = tetra-nor-triterpenoid) – Intense synhtetic efforts by the groups of Nicolaou, Watanabe, Ley and others since structural elucidation in 1987. –1st total synthesis achieved in 2007 by Ley following 22 yrs of effort – ~40 researchers and over 100 person-years of research! – 64-step synthesis – Veitch Angew. Chem. Int. Ed. 2007, 46, 7629 (DOI) & Veitch Angew. Chem. Int. Ed. 2007, 46, 7633 (DOI) – Review ‘The azadirachtin story’ see: Veitch Angew. Chem. Int. Ed. 2008, 47, 9402 (DOI) Format & Scope of Presentation • Metabolism & Biosynthesis – some definitions, 1° & 2° metabolites • Shikimate Metabolites – photosynthesis & glycolysis → shikimate formation → shikimate metabolites – Glyphosate – a non-selective herbicide • Alkaloids – acetylCoA & the citric acid cycle → -amino acids → alkaloids – Opioids – powerful pain killers • Fatty Acids and Polyketides –acetylCoA → malonylCoA → fatty acids, prostaglandins, polyketides, macrolide antibiotics – NSAIDs – anti-inflammatory’s • Isoprenoids/terpenes -

Product Profiles of Egyptian Henbane Premnaspirodiene
The Journal of Antibiotics (2016) 69, 524–533 & 2016 Japan Antibiotics Research Association All rights reserved 0021-8820/16 www.nature.com/ja ORIGINAL ARTICLE Biosynthetic potential of sesquiterpene synthases: product profiles of Egyptian Henbane premnaspirodiene synthase and related mutants Hyun Jo Koo1,3, Christopher R Vickery1,2,3,YiXu1, Gordon V Louie1, Paul E O'Maille1, Marianne Bowman1, Charisse M Nartey1, Michael D Burkart2 and Joseph P Noel1 The plant terpene synthase (TPS) family is responsible for the biosynthesis of a variety of terpenoid natural products possessing diverse biological functions. TPSs catalyze the ionization and, most commonly, rearrangement and cyclization of prenyl diphosphate substrates, forming linear and cyclic hydrocarbons. Moreover, a single TPS often produces several minor products in addition to a dominant product. We characterized the catalytic profiles of Hyoscyamus muticus premnaspirodiene synthase (HPS) and compared it with the profile of a closely related TPS, Nicotiana tabacum 5-epi-aristolochene synthase (TEAS). The profiles of two previously studied HPS and TEAS mutants, each containing nine interconverting mutations, dubbed HPS-M9 and TEAS- M9, were also characterized. All four TPSs were compared under varying temperature and pH conditions. In addition, we solved the X-ray crystal structures of TEAS and a TEAS quadruple mutant complexed with substrate and products to gain insight into the enzymatic features modulating product formation. These informative structures, along with product profiles, -

Toxina PR En Penicillium Roqueforti
Universidad de León Instituto de Biotecnología de León Dpto. de Biología Molecular INBIOTEC Área de Microbiología TESIS DOCTORAL Caracterización Bioquímica y Molecular de la Biosíntesis del Antitumoral Andrastina y de la Toxina PR en Penicillium roqueforti . Pedro Iván Hidalgo Yanes León 2013 INFORME DEL DIRECTOR DE LA TESIS 1 (R.D. 99/2011, de 28 de enero y Normativa de la ULE) El Dr. D. Juan Francisco Martín Martín, el Dr. D. Ricardo Vicente Ullán y la Dra. Dña. Silvia Albillos García como Directores 2 de la Tesis Doctoral titulada “Caracterización Bioquímica y Molecular de la Biosíntesis del Antitumoral Andrastina y de la Toxina PR en Penicillium roqueforti ” realizada por D. Pedro Iván Hidalgo Yanes en el programa de doctorado Biología Molecular y Biotecnología, informan favorablemente el depósito de la misma, dado que reúne las condiciones necesarias para su defensa. León a ___ de__________ de _____________ D. Juan Francisco Martín Martín D. Ricardo Vicente Ullán Dña. Silvia Albillos García 1 Este impreso solamente se cumplimentará para los casos de tesis depositadas en papel. 2 Si la Tesis está dirigida por más de un Director tienen que constar los datos de cada uno y han de firmar todos ellos. ADMISIÓN A TRÁMITE DE LA TESIS DOCTORAL 3 (R.D. 99/2011, de 28 de enero y Normativa de la ULE) El órgano responsable del programa de doctorado _____________________________ ___________________________________________________________________________ en su reunión celebrada el día ___ de _____________ de ________ ha acordado dar su conformidad a la admisión a trámite de lectura de la Tesis Doctoral titulada “Caracterización Bioquímica y Molecular de la Biosíntesis del Antitumoral Andrastina y de la Toxina PR en Penicillium roqueforti ”, dirigida por el Dr. -

Searching for Novel Targets to Control Wheat Head Blight Disease—I-Protein Identification, 3D Modeling and Virtual Screening
Advances in Microbiology, 2016, 6, 811-830 http://www.scirp.org/journal/aim ISSN Online: 2165-3410 ISSN Print: 2165-3402 Searching for Novel Targets to Control Wheat Head Blight Disease—I-Protein Identification, 3D Modeling and Virtual Screening Natália F. Martins1, Emmanuel Bresso1, Roberto C. Togawa1, Martin Urban2, John Antoniw2, Bernard Maigret3, Kim Hammond-Kosack2 1EMBRAPA Recursos Genéticos e Biotecnologia Parque Estação Biológica, Brasília, Brazil 2Department of Plant Biology and Crop Science, Rothamsted Research, Harpenden, UK 3CNRS, LORIA, UMR 7503, Lorraine University, Nancy, France How to cite this paper: Martins, N.F., Abstract Bresso, E., Togawa, R.C., Urban, M., Anto- niw, J., Maigret, B. and Hammond-Kosack, Fusarium head blight (FHB) is a destructive disease of wheat and other cereals. FHB K. (2016) Searching for Novel Targets to occurs in Europe, North America and around the world causing significant losses in Control Wheat Head Blight Disease— production and endangers human and animal health. In this article, we provide the I-Protein Identification, 3D Modeling and strategic steps for the specific target selection for the phytopathogen system wheat- Virtual Screening. Advances in Microbiol- ogy, 6, 811-830. Fusarium graminearum. The economic impact of FHB leads to the need for innova- http://dx.doi.org/10.4236/aim.2016.611079 tion. Currently used fungicides have been shown to be effective over the years, but recently cereal infecting Fusaria have developed resistance. Our work presents a new Received: June 21, 2016 perspective on target selection to allow the development of new fungicides. We de- Accepted: September 11, 2016 veloped an innovative approach combining both genomic analysis and molecular Published: September 14, 2016 modeling to increase the discovery for new chemical compounds with both safety Copyright © 2016 by authors and and low environmental impact. -

Biology Chemistry III: Computers in Education High School
Abstracts 1-68 Relate to the Sunday Program Biology 1. 100 Years of Genetics William Sofer, Rutgers University, Piscataway, NJ Almost exactly 100 years ago, Thomas Hunt Morgan and his coworkers at Columbia University began studying a small fly, Drosophila melanogaster, in an effort to learn something about the laws of heredity. After a while, they found a single white-eyed male among many thousands of normal red-eyed males and females. The analysis of the offspring that resulted from crossing this mutant male with red-eyed females led the way to the discovery of what determines whether an individual becomes a male or a female, and the relationship of chromosomes and genes. 2. Streptomycin - Antibiotics from the Ground Up Douglas Eveleigh, Rutgers University, New Brunswick, NJ Antibiotics are part of everyday living. We benefit from their use through prevention of infection of cuts and scratches, control of diseases such as typhoid, cholera and potentially of bioterrorist's pathogens, besides allowing the marvels of complex surgeries. Antibiotics are a wondrous medical weapon. But where do they come from? The unlikely answer is soil. Soil is home to a teeming population of insects and roots, plus billions of microbes - billions. But life is not harmonious in soil. Some microbes have evolved strategies to dominate their territory; one strategem is the production of antibiotics. In the 1940s, Selman Waksman, with his research team at Rutgers University, began the first ever search for such antibiotic producing micro-organisms amidst the thousands of soil microbes. The first antibiotics they discovered killed microbes but were toxic to humans. -

JPRI Working Paper No. 3: October 1994 Strengths and Weaknesses of Education in Japan by Masao Kunihiro Academic Apartheid at Ja
JPRI Working Paper No. 3: October 1994 Strengths and Weaknesses of Education in Japan By Masao Kunihiro Academic Apartheid at Japan’s National Universities By Ivan P. Hall Strengths and Weaknesses of Education in Japan By Masao Kunihiro I am not, by any stretch of semantic generosity, an expert in either pedagogy or the sociology of knowledge. However, I have been associated with several universities as a teacher, and have published several books pertaining to education in the broad sense of the term, including translations into Japanese of David Riesman’s The Academic Revolution, Herbert Passin’s Society and Education in Japan, and Benjamin Duke’s The Japanese School. That is to say, I am interested in education as a human endeavor in general, and education in Japan in particular. I have also been associated with the educational end of NHK television and sat on the Education Committee in the Upper House of the Diet for a total of four years. Let me begin with some brief comments about the Sinitic Culture Area--of which Japan is a part--and the possible impact of Confucianism on education. According to Ronald Dore’s 1989 speech, “Confucianism, Economic Growth and Social Development,” there are four salient characteristics of Confucianism relevant to education. First: dutifulness to a larger collectivity, as opposed to individual rights to the pursuit of happiness. Second: the proclivity toward accepting a system of hierarchy. Third: special roles assigned to elites who are highly educated; those with knowledge are entitled to moral authority to rule. Fourth: rationality. Professor De Bary of Columbia University has maintained that, popular views of Confucianism as authoritarian to the contrary, a case can readily be made for both Liberalism and Democracy within the Confucian tradition. -

Electric Supporting Information (ESI) Crystal Structure and Functional
Electronic Supplementary Material (ESI) for Chemical Science. This journal is © The Royal Society of Chemistry 2018 Electric Supporting Information (ESI) Crystal structure and functional analysis of large-terpene synthase belonging to a newly found subclass Masahiro Fujihashi,a* Tsutomu Sato,b* Yuma Tanaka,a Daisuke Yamamoto,a Tomoyuki Nishi,b Daijiro Ueda,b Mizuki Murakami,b Yoko Yasuno,c Ai Sekihara,c Kazuma Fuku,c Tetsuro Shinadac & Kunio Mikia* a. Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: [email protected], [email protected] b. Department of Applied Biological Chemistry, Faculty of Agriculture, and Graduate School of Science and Technology, Niigata University, 8050 Ikarashi-2, Niigata 950-2181, Japan. E-mail: [email protected] c. Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi, Osaka 558-8585, Japan. S1 Table of Contents Experimental Procedures S3 General procedure S3 Vector construction, expression and purification of BalTS S3 Crystallization and crystallographic analysis of balTS S3 Oligomeric state analysis S4 Analysis of the dimer geometry S4 Vector construction, expression and purification of BsuTS S4 Homology modeling of BsuTS S4 Enzymatic assays S5 Isolation and identification of β-springen S5 Movie Captions S6 Author Contributions S6 Figures Fig. S1 S7 Fig. S2 S8 Fig. S3 S9 Fig. S4 S10 Fig. S5 S17 Fig. S6 S18 Fig. S7, S8 S19 Fig. S9 S20 Fig. S10 S21 Fig. S11 S22 Tables Table S1 S23 Table S2 S24 References S27 S2 Experimental Procedures General procedure NMR spectra were recorded using a Bruker DPX 400 spectrometer at 400 MHz for proton (1H) and 100 MHz for carbon (13C).