A Genomics-Led Approach to Deciphering Heterocyclic Natural
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METACYC ID Description A0AR23 GO:0004842 (Ubiquitin-Protein Ligase
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Heat Stress Responsive Zostera marina Genes, Southern Population (α=0. -

Structure-Based Virtual Screening of Hypothetical Inhibitors of the Enzyme Longiborneol Synthase—A Potential Target to Reduce Fusarium Head Blight Disease
J Mol Model (2016) 22: 163 DOI 10.1007/s00894-016-3021-1 ORIGINAL PAPER Structure-based virtual screening of hypothetical inhibitors of the enzyme longiborneol synthase—a potential target to reduce Fusarium head blight disease E. Bresso1 & V. L ero ux 2 & M. Urban3 & K. E. Hammond-Kosack3 & B. Maigret2 & N. F. Martins1 Received: 17 December 2015 /Accepted: 27 May 2016 /Published online: 21 June 2016 # Springer-Verlag Berlin Heidelberg 2016 Abstract Fusarium head blight (FHB) is one of the most compounds from a library of 15,000 drug-like compounds. destructive diseases of wheat and other cereals worldwide. These putative inhibitors of longiborneol synthase provide a During infection, the Fusarium fungi produce mycotoxins that sound starting point for further studies involving molecular represent a high risk to human and animal health. Developing modeling coupled to biochemical experiments. This process small-molecule inhibitors to specifically reduce mycotoxin could eventually lead to the development of novel approaches levels would be highly beneficial since current treatments to reduce mycotoxin contamination in harvested grain. unspecifically target the Fusarium pathogen. Culmorin pos- sesses a well-known important synergistically virulence role Keywords Fusarium mycotoxins . Culmorin . Inhibitors . among mycotoxins, and longiborneol synthase appears to be a Homology modeling . Molecular dynamics . Ensemble key enzyme for its synthesis, thus making longiborneol syn- docking thase a particularly interesting target. This study aims to dis- cover potent and less toxic agrochemicals against FHB. These compounds would hamper culmorin synthesis by inhibiting Introduction longiborneol synthase. In order to select starting molecules for further investigation, we have conducted a structure- Fusarium head blight (FHB), caused by Fusarium based virtual screening investigation. -

Intraprotein Radical Transfer During Photoactivation of DNA Photolyase
letters to nature tri-NaCitrate and 20% PEG 3350, at a ®nal pH of 7.4 (PEG/Ion Screen, Hampton (Daresbury Laboratory, Warrington, 1992). Research, San Diego, California) within two weeks at 4 8C. Intact complex was veri®ed by 25. Evans P. R. in Proceedings of the CCP4 Study Weekend. Data Collection and Processing (eds Sawyer, L., SDS±polyacrylamide gel electrophoresis of washed crystals (see Supplementary Infor- Isaacs, N. & Bailey, S.) 114±122 (Daresbury Laboratory, 1993). mation). Data were collected from a single frozen crystal, cryoprotected in 28.5% PEG 26. Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystal- 4000 and 10% PEG 400, at beamline 9.6 at the SRS Daresbury, UK. lography. Acta Crystallogr. 50, 760±763 (1994). The data were processed using MOSFLM24 and merged using SCALA25 from the CCP4 27. Navaza, J. AMORE - An automated package for molecular replacement. Acta Cryst. A 50, 157±163 26 (1994). package (Table 1) The molecular replacement solution for a1-antitrypsin in the complex 27 28 28. Engh, R. et al. The S variant of human alpha 1-antitrypsin, structure and implications for function and was obtained using AMORE and the structure of cleaved a1-antitrypsin as the search model. Conventional molecular replacement searches failed to place a model of intact metabolism. Protein Eng. 2, 407±415 (1989). 29 29. Lee, S. L. New inhibitors of thrombin and other trypsin-like proteases: hydrogen bonding of an trypsin in the complex, although maps calculated with phases from a1-antitrypsin alone showed clear density for the ordered portion of trypsin (Fig. -

Downloaded 10/5/2021 9:43:05 AM
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Aromatic side-chain flips orchestrate the conformational sampling of functional loops in Cite this: Chem. Sci.,2021,12,9318 † All publication charges for this article human histone deacetylase 8 have been paid for by the Royal Society a a bcd of Chemistry Vaibhav Kumar Shukla, ‡ Lucas Siemons, ‡ Francesco L. Gervasio and D. Flemming Hansen *a Human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and an important drug-target. High-resolution structures of HDAC8 in complex with substrates or inhibitors are available, which have provided insights into the bound state of HDAC8 and its function. Here, using long all-atom unbiased molecular dynamics simulations and Markov state modelling, we show a strong correlation between the conformation of aromatic side chains near the active site and opening and closing of the surrounding functional loops of HDAC8. We also investigated two mutants known to allosterically downregulate the enzymatic activity of HDAC8. Based on experimental data, we hypothesise that I19S-HDAC8 is unable to Received 6th April 2021 Creative Commons Attribution 3.0 Unported Licence. release the product, whereas both product release and substrate binding are impaired in the S39E- Accepted 27th May 2021 HDAC8 mutant. The presented results deliver detailed insights into the functional dynamics of HDAC8 DOI: 10.1039/d1sc01929e and provide a mechanism for the substantial downregulation caused by allosteric mutations, including rsc.li/chemical-science a disease causing one. Introduction II (HDAC-4, -5, -6, -7, -9, and HDAC10), and class III (SIRT1-7) have sequence similarity to yeast Rpd3, Hda1, and Sir2, Acetylation of lysine side chains occurs as a co-translation or respectively, whereas class IV (HDAC11) shares sequence simi- 2 This article is licensed under a post-translational modication of proteins and was rst iden- larity with both class I and II proteins. -

Differentiating Two Closely Related Alexandrium Species Using Comparative Quantitative Proteomics
toxins Article Differentiating Two Closely Related Alexandrium Species Using Comparative Quantitative Proteomics Bryan John J. Subong 1,2,* , Arturo O. Lluisma 1, Rhodora V. Azanza 1 and Lilibeth A. Salvador-Reyes 1,* 1 Marine Science Institute, University of the Philippines- Diliman, Velasquez Street, Quezon City 1101, Philippines; [email protected] (A.O.L.); [email protected] (R.V.A.) 2 Department of Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo City, Tokyo 113-8654, Japan * Correspondence: [email protected] (B.J.J.S.); [email protected] (L.A.S.-R.) Abstract: Alexandrium minutum and Alexandrium tamutum are two closely related harmful algal bloom (HAB)-causing species with different toxicity. Using isobaric tags for relative and absolute quantita- tion (iTRAQ)-based quantitative proteomics and two-dimensional differential gel electrophoresis (2D-DIGE), a comprehensive characterization of the proteomes of A. minutum and A. tamutum was performed to identify the cellular and molecular underpinnings for the dissimilarity between these two species. A total of 1436 proteins and 420 protein spots were identified using iTRAQ-based proteomics and 2D-DIGE, respectively. Both methods revealed little difference (10–12%) between the proteomes of A. minutum and A. tamutum, highlighting that these organisms follow similar cellular and biological processes at the exponential stage. Toxin biosynthetic enzymes were present in both organisms. However, the gonyautoxin-producing A. minutum showed higher levels of osmotic growth proteins, Zn-dependent alcohol dehydrogenase and type-I polyketide synthase compared to the non-toxic A. tamutum. Further, A. tamutum had increased S-adenosylmethionine transferase that may potentially have a negative feedback mechanism to toxin biosynthesis. -

Characterization of a Cytochrome P450 Monooxygenase Gene Involved in the Biosynthesis of Geosmin in Penicillium Expansum
Open Archive TOULOUSE Archive Ouverte (OATAO) OATAO is an open access repository that collects the work of Toulouse researchers and makes it freely available over the web where possible. This is an author-deposited version published in : http://oatao.univ-toulouse.fr/ Eprints ID : 10812 To link to this article : DOI : 10.5897/AJMR11.1361 URL : http://academicjournals.org/journal/AJMR/article- abstract/841ECBA21771 To cite this version : Siddique, Muhammad Hussnain and Liboz, Thierry and Bacha, Nafees and Puel, Olivier and Mathieu, Florence and Lebrihi, Ahmed Characterization of a cytochrome P450 monooxygenase gene involved in the biosynthesis of geosmin in Penicillium expansum. (2012) African Journal of Microbiology Research, vol. 6 (n° 19). pp. 4122-4127. ISSN 1996-0808 Any correspondance concerning this service should be sent to the repository administrator: [email protected] Characterization of a cytochrome P450 monooxygenase gene involved in the biosynthesis of geosmin in Penicillium expansum Muhammad Hussnain Siddique1,2, Thierry Liboz1,2, Nafees Bacha3, Olivier Puel4, Florence Mathieu1,2 and Ahmed Lebrihi1,2,5* 1Université de Toulouse, INPT-UPS, Laboratoire de Génie Chimique, avenue de l’Agrobiopole, 31326 Castanet-Tolosan Cedex, France. 2Le Centre national de la recherche scientifique (CNRS), Laboratoire de Génie Chimique, 31030 Toulouse, France. 3Centre of Biotechnology and Microbiology, University of Peshawar, Pakistan. 4Institut National de la Recherche Agronomique (INRA), Laboratoire de Pharmacologie Toxicologie, 31931 Toulouse, France. 5Université Moulay Ismail, Marjane 2, BP 298, Meknes, Morocco. Geosmin is a terpenoid, an earthy-smelling substance associated with off-flavors in water and wine. The biosynthesis of geosmin is well characterized in bacteria, but little is known about its production in eukaryotes, especially in filamentous fungi. -

N-Carbamoylation of 2,4-Diaminobutyrate Reroutes the Outcome in Padanamide Biosynthesis
Chemistry & Biology Article N-Carbamoylation of 2,4-Diaminobutyrate Reroutes the Outcome in Padanamide Biosynthesis Yi-Ling Du,1 Doralyn S. Dalisay,1 Raymond J. Andersen,1,2 and Katherine S. Ryan1,* 1Department of Chemistry 2Department of Earth, Ocean and Atmospheric Sciences University of British Columbia, Vancouver, BC V6T 1Z1, Canada *Correspondence: [email protected] http://dx.doi.org/10.1016/j.chembiol.2013.06.013 SUMMARY literature. It is interesting that a compound identical to padana- mide A, named actinoramide A (Nam et al., 2011), was indepen- Padanamides are linear tetrapeptides notable for the dently reported from Streptomyces sp. CNQ-027. This actinomy- absence of proteinogenic amino acids in their struc- cete strain was isolated from sediment on the opposite side of the tures. In particular, two unusual heterocycles, (S)- Pacific Ocean, near San Diego, CA, suggesting a potentially wide 3-amino-2-oxopyrrolidine-1-carboxamide (S-Aopc) distribution of the padanamides/actinoramides. It is intriguing and (S)-3-aminopiperidine-2,6-dione (S-Apd), are that, whereas padanamide A and actinoramide A are identical, found at the C-termini of padanamides A and B, the minor compounds (actinoramides B and C) co-isolated from Streptomyces sp. CNQ-027 are unique (Figure 1A). Total respectively. Here we identify the padanamide synthesis of padanamides A and B was recently reported (Long biosynthetic gene cluster and carry out systematic et al., 2013), confirming the previous structural elucidations. gene inactivation studies. Our results show that The padanamides attracted our attention for their many padanamides are synthesized by highly dissociated unusual chemical features. -

Development of a Phage Display Library for Discovery of Antigenic Brucella Peptides Jeffrey Williams Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2018 Development of a phage display library for discovery of antigenic Brucella peptides Jeffrey Williams Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Microbiology Commons Recommended Citation Williams, Jeffrey, "Development of a phage display library for discovery of antigenic Brucella peptides" (2018). Graduate Theses and Dissertations. 16896. https://lib.dr.iastate.edu/etd/16896 This Thesis is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Development of a phage display library for discovery of antigenic Brucella peptides by Jeffrey Williams A thesis submitted to the graduate faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Major: Microbiology Program of Study Committee: Bryan H. Bellaire, Major Professor Steven Olsen Steven Carlson The student author, whose presentation of the scholarship herein was approved by the program of study committee, is solely responsible for the content of this thesis. The Graduate College will ensure this thesis is globally accessible and will not permit alterations after a degree is conferred. Iowa State University -
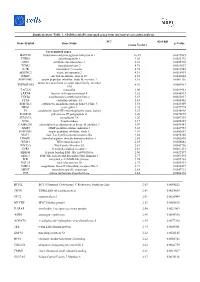
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Metatranscriptomic Analysis of Community Structure And
School of Environmental Sciences Metatranscriptomic analysis of community structure and metabolism of the rhizosphere microbiome by Thomas Richard Turner Submitted in partial fulfilment of the requirement for the degree of Doctor of Philosophy, September 2013 This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with the author and that use of any information derived there from must be in accordance with current UK Copyright Law. In addition, any quotation or extract must include full attribution. i Declaration I declare that this is an account of my own research and has not been submitted for a degree at any other university. The use of material from other sources has been properly and fully acknowledged, where appropriate. Thomas Richard Turner ii Acknowledgements I would like to thank my supervisors, Phil Poole and Alastair Grant, for their continued support and guidance over the past four years. I’m grateful to all members of my lab, both past and present, for advice and friendship. Graham Hood, I don’t know how we put up with each other, but I don’t think I could have done this without you. Cheers Salt! KK, thank you for all your help in the lab, and for Uma’s biryanis! Andrzej Tkatcz, thanks for the useful discussions about our projects. Alison East, thank you for all your support, particularly ensuring Graham and I did not kill each other. I’m grateful to Allan Downie and Colin Murrell for advice. For sequencing support, I’d like to thank TGAC, particularly Darren Heavens, Sophie Janacek, Kirsten McKlay and Melanie Febrer, as well as John Walshaw, Mark Alston and David Swarbreck for bioinformatic support. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

Description of Streptomyces Dangxiongensis Sp. Nov
Cronfa - Swansea University Open Access Repository _____________________________________________________________ This is an author produced version of a paper published in: International Journal of Systematic and Evolutionary Microbiology Cronfa URL for this paper: http://cronfa.swan.ac.uk/Record/cronfa50961 _____________________________________________________________ Paper: Zhang, B., Tang, S., Yang, R., Chen, X., Zhang, D., Zhang, W., Li, S., Chen, T., Liu, G. et. al. (2019). Streptomyces dangxiongensis sp. nov., isolated from soil of Qinghai-Tibet Plateau. International Journal of Systematic and Evolutionary Microbiology http://dx.doi.org/10.1099/ijsem.0.003550 _____________________________________________________________ This item is brought to you by Swansea University. Any person downloading material is agreeing to abide by the terms of the repository licence. Copies of full text items may be used or reproduced in any format or medium, without prior permission for personal research or study, educational or non-commercial purposes only. The copyright for any work remains with the original author unless otherwise specified. The full-text must not be sold in any format or medium without the formal permission of the copyright holder. Permission for multiple reproductions should be obtained from the original author. Authors are personally responsible for adhering to copyright and publisher restrictions when uploading content to the repository. http://www.swansea.ac.uk/library/researchsupport/ris-support/ Streptomyces dangxiongensis sp. nov., isolated from soil of Qinghai-Tibet Plateau Binglin Zhang1,2,3, Shukun Tang4, Ruiqi Yang1,3, Ximing Chen1,3, Dongming Zhang1, Wei Zhang1,3, Shiweng Li5, Tuo Chen2,3, Guangxiu Liu1,3, Paul Dyson6 1 Key Laboratory of Desert and Desertification, Northwest Institute of Eco-Environment and Resources, Chinese Academy of Sciences, Lanzhou 730000, China.