Introduction to Enzyme and Coenzyme Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Regulation of Plant Monoterpene Biosynthesis in Relation to Fragrance
Molecular Regulation of Plant Monoterpene Biosynthesis In Relation To Fragrance Mazen K. El Tamer Promotor: Prof. Dr. A.G.J Voragen, hoogleraar in de Levensmiddelenchemie, Wageningen Universiteit Co-promotoren: Dr. ir. H.J Bouwmeester, senior onderzoeker, Business Unit Celcybernetica, Plant Research International Dr. ir. J.P Roozen, departement Agrotechnologie en Voedingswetenschappen, Wageningen Universiteit Promotiecommissie: Dr. M.C.R Franssen, Wageningen Universiteit Prof. Dr. J.H.A Kroeze, Wageningen Universiteit Prof. Dr. A.J van Tunen, Swammerdam Institute for Life Sciences, Universiteit van Amsterdam. Prof. Dr. R.G.F Visser, Wageningen Universiteit Mazen K. El Tamer Molecular Regulation Of Plant Monoterpene Biosynthesis In Relation To Fragrance Proefschrift ter verkrijging van de graad van doctor op gezag van de rector magnificus van Wageningen Universiteit, Prof. dr. ir. L. Speelman, in het openbaar te verdedigen op woensdag 27 november 2002 des namiddags te vier uur in de Aula Mazen K. El Tamer Molecular Regulation Of Plant Monoterpene Biosynthesis In Relation To Fragrance Proefschrift Wageningen Universiteit ISBN 90-5808-752-2 Cover and Invitation Design: Zeina K. El Tamer This thesis is dedicated to my Family & Friends Contents Abbreviations Chapter 1 General introduction and scope of the thesis 1 Chapter 2 Monoterpene biosynthesis in lemon (Citrus limon) cDNA isolation 21 and functional analysis of four monoterpene synthases Chapter 3 Domain swapping of Citrus limon monoterpene synthases: Impact 57 on enzymatic activity and -

Structure-Based Virtual Screening of Hypothetical Inhibitors of the Enzyme Longiborneol Synthase—A Potential Target to Reduce Fusarium Head Blight Disease
J Mol Model (2016) 22: 163 DOI 10.1007/s00894-016-3021-1 ORIGINAL PAPER Structure-based virtual screening of hypothetical inhibitors of the enzyme longiborneol synthase—a potential target to reduce Fusarium head blight disease E. Bresso1 & V. L ero ux 2 & M. Urban3 & K. E. Hammond-Kosack3 & B. Maigret2 & N. F. Martins1 Received: 17 December 2015 /Accepted: 27 May 2016 /Published online: 21 June 2016 # Springer-Verlag Berlin Heidelberg 2016 Abstract Fusarium head blight (FHB) is one of the most compounds from a library of 15,000 drug-like compounds. destructive diseases of wheat and other cereals worldwide. These putative inhibitors of longiborneol synthase provide a During infection, the Fusarium fungi produce mycotoxins that sound starting point for further studies involving molecular represent a high risk to human and animal health. Developing modeling coupled to biochemical experiments. This process small-molecule inhibitors to specifically reduce mycotoxin could eventually lead to the development of novel approaches levels would be highly beneficial since current treatments to reduce mycotoxin contamination in harvested grain. unspecifically target the Fusarium pathogen. Culmorin pos- sesses a well-known important synergistically virulence role Keywords Fusarium mycotoxins . Culmorin . Inhibitors . among mycotoxins, and longiborneol synthase appears to be a Homology modeling . Molecular dynamics . Ensemble key enzyme for its synthesis, thus making longiborneol syn- docking thase a particularly interesting target. This study aims to dis- cover potent and less toxic agrochemicals against FHB. These compounds would hamper culmorin synthesis by inhibiting Introduction longiborneol synthase. In order to select starting molecules for further investigation, we have conducted a structure- Fusarium head blight (FHB), caused by Fusarium based virtual screening investigation. -

Rehmannia Glutinosa-Monocultured Rhizosphere Soil
Comparative Metaproteomic Analysis on Consecutively Rehmannia glutinosa-Monocultured Rhizosphere Soil Linkun Wu1,2, Haibin Wang1,2., Zhixing Zhang1,2., Rui Lin2,3, Zhongyi Zhang1,4, Wenxiong Lin1,2* 1 School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2 Agroecological Institute, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 3 College of Oceanography and Environmental Science, Xiamen University, Xiamen, Fujian, China, 4 Institute of Chinese Medicinal Materials, Henan Agriculture University, Zhengzhou, Henan, China Abstract Background: The consecutive monoculture for most of medicinal plants, such as Rehmannia glutinosa, results in a significant reduction in the yield and quality. There is an urgent need to study for the sustainable development of Chinese herbaceous medicine. Methodology/Principal Findings: Comparative metaproteomics of rhizosphere soil was developed and used to analyze the underlying mechanism of the consecutive monoculture problems of R. glutinosa. The 2D-gel patterns of protein spots for the soil samples showed a strong matrix dependency. Among the spots, 103 spots with high resolution and repeatability were randomly selected and successfully identified by MALDI TOF-TOF MS for a rhizosphere soil metaproteomic profile analysis. These proteins originating from plants and microorganisms play important roles in nutrient cycles and energy flow in rhizospheric soil ecosystem. They function in protein, nucleotide and secondary metabolisms, signal transduction and resistance. Comparative metaproteomics analysis revealed 33 differentially expressed protein spots in rhizosphere soil in response to increasing years of monoculture. Among them, plant proteins related to carbon and nitrogen metabolism and stress response, were mostly up-regulated except a down-regulated protein (glutathione S-transferase) involving detoxification. -
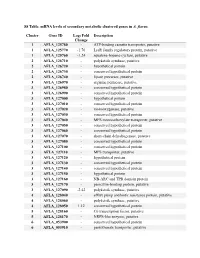
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

(12) United States Patent (10) Patent No.: US 9,115,366 B2 Tissier Et Al
USOO9115366B2 (12) United States Patent (10) Patent No.: US 9,115,366 B2 Tissier et al. (45) Date of Patent: *Aug. 25, 2015 (54) SYSTEM FOR PRODUCING TERPENOIDS IN WO WO99,38957 * 8, 1999 .......... C12N 15/82 PLANTS WO WO99,38957 A1 8/1999 WO WOOOf 17327 A3 3.2000 Fre WO WO 01/20008 A2 3, 2001 (75) Inventors: Alain Tissier, Pertuis (FR); Christophe WO WO 2004/111183 A2 12/2004 Sallaud, Montpellier (FR); Denis WO WO 2006/04.0479 4/2006 Rontein,ontein, GreouxG les Bains (FR(FR) OTHER PUBLICATIONS (73) Assignee: PHILIP MORRIS PRODUCTS S.A., Aharoni, Aetal. The Plant Cell (Dec. 2003), vol. 15: pp. 2866-2884.* Neuchatel (CH) Besumbes, O. et al. Biotechnology and Bioengineering; Oct. 20. 2004; vol. 88, No. 2: pp. 168-175.* (*) Notice: Subject to any disclaimer, the term of this Wang, E. et al. Nature Biotechnology, Apr. 2001; vol. 19, pp. 371 patent is extended or adjusted under 35 37.4% U.S.C. 154(b) by 900 days. Wang, E. et al. Journal of Experimental Botany, Sep. 2002, vol. 53, No. 376; pp. 1891-1897.* This patent is Subject to a terminal dis Walker K. et al. Phytochemistry (2001) vol. 58; pp. 1-7.* claimer. Gutiérrez-Alcalá et al., A versatile promoter for the expression of proteins in glandular and non-glandular trichomes from a variety of plants, 56 J of Exp Botany No. 419, 2487-2494 (2005).* (21) Appl. No.: 11/814,943 Besumbes et al. (Metabolic Engineering of Isoprenoid Biosynthesis in Arabidopsis for the Production of Taxadiene, the First Committed (22) PCT Filed: Jan. -

Searching for Novel Targets to Control Wheat Head Blight Disease—I-Protein Identification, 3D Modeling and Virtual Screening
Advances in Microbiology, 2016, 6, 811-830 http://www.scirp.org/journal/aim ISSN Online: 2165-3410 ISSN Print: 2165-3402 Searching for Novel Targets to Control Wheat Head Blight Disease—I-Protein Identification, 3D Modeling and Virtual Screening Natália F. Martins1, Emmanuel Bresso1, Roberto C. Togawa1, Martin Urban2, John Antoniw2, Bernard Maigret3, Kim Hammond-Kosack2 1EMBRAPA Recursos Genéticos e Biotecnologia Parque Estação Biológica, Brasília, Brazil 2Department of Plant Biology and Crop Science, Rothamsted Research, Harpenden, UK 3CNRS, LORIA, UMR 7503, Lorraine University, Nancy, France How to cite this paper: Martins, N.F., Abstract Bresso, E., Togawa, R.C., Urban, M., Anto- niw, J., Maigret, B. and Hammond-Kosack, Fusarium head blight (FHB) is a destructive disease of wheat and other cereals. FHB K. (2016) Searching for Novel Targets to occurs in Europe, North America and around the world causing significant losses in Control Wheat Head Blight Disease— production and endangers human and animal health. In this article, we provide the I-Protein Identification, 3D Modeling and strategic steps for the specific target selection for the phytopathogen system wheat- Virtual Screening. Advances in Microbiol- ogy, 6, 811-830. Fusarium graminearum. The economic impact of FHB leads to the need for innova- http://dx.doi.org/10.4236/aim.2016.611079 tion. Currently used fungicides have been shown to be effective over the years, but recently cereal infecting Fusaria have developed resistance. Our work presents a new Received: June 21, 2016 perspective on target selection to allow the development of new fungicides. We de- Accepted: September 11, 2016 veloped an innovative approach combining both genomic analysis and molecular Published: September 14, 2016 modeling to increase the discovery for new chemical compounds with both safety Copyright © 2016 by authors and and low environmental impact. -

Electric Supporting Information (ESI) Crystal Structure and Functional
Electronic Supplementary Material (ESI) for Chemical Science. This journal is © The Royal Society of Chemistry 2018 Electric Supporting Information (ESI) Crystal structure and functional analysis of large-terpene synthase belonging to a newly found subclass Masahiro Fujihashi,a* Tsutomu Sato,b* Yuma Tanaka,a Daisuke Yamamoto,a Tomoyuki Nishi,b Daijiro Ueda,b Mizuki Murakami,b Yoko Yasuno,c Ai Sekihara,c Kazuma Fuku,c Tetsuro Shinadac & Kunio Mikia* a. Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: [email protected], [email protected] b. Department of Applied Biological Chemistry, Faculty of Agriculture, and Graduate School of Science and Technology, Niigata University, 8050 Ikarashi-2, Niigata 950-2181, Japan. E-mail: [email protected] c. Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi, Osaka 558-8585, Japan. S1 Table of Contents Experimental Procedures S3 General procedure S3 Vector construction, expression and purification of BalTS S3 Crystallization and crystallographic analysis of balTS S3 Oligomeric state analysis S4 Analysis of the dimer geometry S4 Vector construction, expression and purification of BsuTS S4 Homology modeling of BsuTS S4 Enzymatic assays S5 Isolation and identification of β-springen S5 Movie Captions S6 Author Contributions S6 Figures Fig. S1 S7 Fig. S2 S8 Fig. S3 S9 Fig. S4 S10 Fig. S5 S17 Fig. S6 S18 Fig. S7, S8 S19 Fig. S9 S20 Fig. S10 S21 Fig. S11 S22 Tables Table S1 S23 Table S2 S24 References S27 S2 Experimental Procedures General procedure NMR spectra were recorded using a Bruker DPX 400 spectrometer at 400 MHz for proton (1H) and 100 MHz for carbon (13C). -

(10) Patent No.: US 8119385 B2
US008119385B2 (12) United States Patent (10) Patent No.: US 8,119,385 B2 Mathur et al. (45) Date of Patent: Feb. 21, 2012 (54) NUCLEICACIDS AND PROTEINS AND (52) U.S. Cl. ........................................ 435/212:530/350 METHODS FOR MAKING AND USING THEMI (58) Field of Classification Search ........................ None (75) Inventors: Eric J. Mathur, San Diego, CA (US); See application file for complete search history. Cathy Chang, San Diego, CA (US) (56) References Cited (73) Assignee: BP Corporation North America Inc., Houston, TX (US) OTHER PUBLICATIONS c Mount, Bioinformatics, Cold Spring Harbor Press, Cold Spring Har (*) Notice: Subject to any disclaimer, the term of this bor New York, 2001, pp. 382-393.* patent is extended or adjusted under 35 Spencer et al., “Whole-Genome Sequence Variation among Multiple U.S.C. 154(b) by 689 days. Isolates of Pseudomonas aeruginosa” J. Bacteriol. (2003) 185: 1316 1325. (21) Appl. No.: 11/817,403 Database Sequence GenBank Accession No. BZ569932 Dec. 17. 1-1. 2002. (22) PCT Fled: Mar. 3, 2006 Omiecinski et al., “Epoxide Hydrolase-Polymorphism and role in (86). PCT No.: PCT/US2OO6/OOT642 toxicology” Toxicol. Lett. (2000) 1.12: 365-370. S371 (c)(1), * cited by examiner (2), (4) Date: May 7, 2008 Primary Examiner — James Martinell (87) PCT Pub. No.: WO2006/096527 (74) Attorney, Agent, or Firm — Kalim S. Fuzail PCT Pub. Date: Sep. 14, 2006 (57) ABSTRACT (65) Prior Publication Data The invention provides polypeptides, including enzymes, structural proteins and binding proteins, polynucleotides US 201O/OO11456A1 Jan. 14, 2010 encoding these polypeptides, and methods of making and using these polynucleotides and polypeptides. -

PURIFICATION of the NATIVE ENZYME and CLONING .AND CHARACTERIZATION of a Cdna for (+ )-6-CADINENE SYNTHASE from BACTERIA-INOCULATED COTTON FOLIAR TISSUE
PURIFICATION OF THE NATIVE ENZYME AND CLONING .AND CHARACTERIZATION OF A cDNA FOR (+ )-6-CADINENE SYNTHASE FROM BACTERIA-INOCULATED COTTON FOLIAR TISSUE By EDWARD M. DAVIS Bachelor of Science Oklahoma State University Stillwater, Oklahoma 1987 Submitted to the Faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY May, 1998 PURIFICATION OF THENATIVE ENZYME AND . CLONING AND CHARACTERIZATIQNOFA cDNA FOR (+ )-B-CADINENE SYNTHASE FROM BACTERIA-INOCULATED COTTON FOLIAR TISSUE Thesis Approved: ~··. L) .. ·g pJ ~fthe Graduate College . · · ii ACKNOWLEDGMENTS I would like to express my appreciation to the faculty, staff and graduate students of the Department of Biochemistry and Molecular Biology who have given both time and energy to assist in my scientific training and development. I would like to thank Margaret and Marlee for giving me the opportunity to participate on a project that includes protein and molecular biological methods. I wish to acknowledge the members of my committee for their time, guidance, and support. I would like to acknowledge Phillips 66 Corporation for their generous donation of equipment, the McAlester Scottish Rite Foundation and the OSU Foundation for financial support, and the EPSCOR program,. the NSF, and the USDA for providing the grants which made this work possible. A special thanks to Drs. Blair, Leach, Melcher, Sensharma and Mitchell for helping to maintain a nearly steady salary when the grant money was not available and to Drs. Cushman and Melcher and Janet Rogers fortechnical support. I would like to thank Drs. Gordon Davis and Steve Hartson for their encouragement and scientific advice, Dr. -

Supporting Information High-Throughput Virtual Screening
Supporting Information High-Throughput Virtual Screening of Proteins using GRID Molecular Interaction Fields Simone Sciabola, Robert V. Stanton, James E. Mills, Maria M. Flocco, Massimo Baroni, Gabriele Cruciani, Francesca Perruccio and Jonathan S. Mason Contents Table S1 S2-S21 Figure S1 S22 * To whom correspondence should be addressed: Simone Sciabola, Pfizer Research Technology Center, Cambridge, 02139 MA, USA Phone: +1-617-551-3327; Fax: +1-617-551-3117; E-mail: [email protected] S1 Table S1. Description of the 990 proteins used as decoy for the Protein Virtual Screening analysis. PDB ID Protein family Molecule Res. (Å) 1n24 ISOMERASE (+)-BORNYL DIPHOSPHATE SYNTHASE 2.3 1g4h HYDROLASE 1,3,4,6-TETRACHLORO-1,4-CYCLOHEXADIENE HYDROLASE 1.8 1cel HYDROLASE(O-GLYCOSYL) 1,4-BETA-D-GLUCAN CELLOBIOHYDROLASE I 1.8 1vyf TRANSPORT PROTEIN 14 KDA FATTY ACID BINDING PROTEIN 1.85 1o9f PROTEIN-BINDING 14-3-3-LIKE PROTEIN C 2.7 1t1s OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.4 1t1r OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.3 1q0q OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 1.9 1jcy LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.9 1fww LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.85 1uk7 HYDROLASE 2-HYDROXY-6-OXO-7-METHYLOCTA-2,4-DIENOATE 1.7 1v11 OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.95 1x7w OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.73 1d0l TRANSFERASE 35KD SOLUBLE LYTIC TRANSGLYCOSYLASE 1.97 2bt4 LYASE 3-DEHYDROQUINATE DEHYDRATASE -

Medically Useful Plant Terpenoids: Biosynthesis, Occurrence, and Mechanism of Action
molecules Review Medically Useful Plant Terpenoids: Biosynthesis, Occurrence, and Mechanism of Action Matthew E. Bergman 1 , Benjamin Davis 1 and Michael A. Phillips 1,2,* 1 Department of Cellular and Systems Biology, University of Toronto, Toronto, ON M5S 3G5, Canada; [email protected] (M.E.B.); [email protected] (B.D.) 2 Department of Biology, University of Toronto–Mississauga, Mississauga, ON L5L 1C6, Canada * Correspondence: [email protected]; Tel.: +1-905-569-4848 Academic Editors: Ewa Swiezewska, Liliana Surmacz and Bernhard Loll Received: 3 October 2019; Accepted: 30 October 2019; Published: 1 November 2019 Abstract: Specialized plant terpenoids have found fortuitous uses in medicine due to their evolutionary and biochemical selection for biological activity in animals. However, these highly functionalized natural products are produced through complex biosynthetic pathways for which we have a complete understanding in only a few cases. Here we review some of the most effective and promising plant terpenoids that are currently used in medicine and medical research and provide updates on their biosynthesis, natural occurrence, and mechanism of action in the body. This includes pharmacologically useful plastidic terpenoids such as p-menthane monoterpenoids, cannabinoids, paclitaxel (taxol®), and ingenol mebutate which are derived from the 2-C-methyl-d-erythritol-4-phosphate (MEP) pathway, as well as cytosolic terpenoids such as thapsigargin and artemisinin produced through the mevalonate (MVA) pathway. We further provide a review of the MEP and MVA precursor pathways which supply the carbon skeletons for the downstream transformations yielding these medically significant natural products. Keywords: isoprenoids; plant natural products; terpenoid biosynthesis; medicinal plants; terpene synthases; cytochrome P450s 1. -

Subject Index Proc
13088 Subject Index Proc. Natl. Acad. Sci. USA 91 (1994) Apoptosis in substantia nigra following developmental striatal excitotoxic Brine shrimp injury, 8117 See Artemia Visualizing hippocampal synaptic function by optical detection of Ca2l Broccol entry through the N-methyl-D-aspartate channel, 8170 See Brassica Amygdala modulation of hippocampal-dependent and caudate Bromophenacyl bromide nucleus-dependent memory processes, 8477 Bromophenacyl bromide binding to the actin-bundling protein I-plastin Distribution of corticotropin-releasing factor receptor mRNA expression inhibits inositol trisphosphate-independent increase in Ca2l in human in the rat brain and pituitary, 8777 neutrophils, 3534 Brownian dynamics Preproenkephalin promoter yields region-specific and long-term Adhesion of hard spheres under the influence of double-layer, van der expression in adult brain after direct in vivo gene transfer via a Waals, and gravitational potentials at a solid/liquid interface, 3004 defective herpes simplex viral vector, 8979 Browsers Intravenous administration of a transferrin receptor antibody-nerve Thorn-like prickles and heterophylly in Cyanea: Adaptations to extinct growth factor conjugate prevents the degeneration of cholinergic avian browsers on Hawaii?, 2810 striatal neurons in a model of Huntington disease, 9077 Bruton agammaglobulinemia Axotomy induces the expression of vasopressin receptors in cranial and Genomic organization and structure of Bruton agammaglobulinemia spinal motor nuclei in the adult rat, 9636 tyrosine kinase: Localization