Localization of GAR Transformylase in Escherichia Coli and Mammalian Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Scale Fitness Profile of Caulobacter Crescentus Grown in Natural Freshwater
Supplemental Material Genome-scale fitness profile of Caulobacter crescentus grown in natural freshwater Kristy L. Hentchel, Leila M. Reyes Ruiz, Aretha Fiebig, Patrick D. Curtis, Maureen L. Coleman, Sean Crosson Tn5 and Tn-Himar: comparing gene essentiality and the effects of gene disruption on fitness across studies A previous analysis of a highly saturated Caulobacter Tn5 transposon library revealed a set of genes that are required for growth in complex PYE medium [1]; approximately 14% of genes in the genome were deemed essential. The total genome insertion coverage was lower in the Himar library described here than in the Tn5 dataset of Christen et al (2011), as Tn-Himar inserts specifically into TA dinucleotide sites (with 67% GC content, TA sites are relatively limited in the Caulobacter genome). Genes for which we failed to detect Tn-Himar insertions (Table S13) were largely consistent with essential genes reported by Christen et al [1], with exceptions likely due to differential coverage of Tn5 versus Tn-Himar mutagenesis and differences in metrics used to define essentiality. A comparison of the essential genes defined by Christen et al and by our Tn5-seq and Tn-Himar fitness studies is presented in Table S4. We have uncovered evidence for gene disruptions that both enhanced or reduced strain fitness in lake water and M2X relative to PYE. Such results are consistent for a number of genes across both the Tn5 and Tn-Himar datasets. Disruption of genes encoding three metabolic enzymes, a class C β-lactamase family protein (CCNA_00255), transaldolase (CCNA_03729), and methylcrotonyl-CoA carboxylase (CCNA_02250), enhanced Caulobacter fitness in Lake Michigan water relative to PYE using both Tn5 and Tn-Himar approaches (Table S7). -

B Number Gene Name Mrna Intensity Mrna Present # of Tryptic
list list sample) short list predicted B number Gene name assignment mRNA present mRNA intensity Gene description Protein detected - Membrane protein detected (total list) detected (long list) membrane sample Proteins detected - detected (short list) # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides Functional category detected (membrane Protein detected - total Protein detected - long b0003 thrB 6781 P 9 P 3 3 P 3 0 homoserine kinase Metabolism of small molecules b0004 thrC 15039 P 18 P 10 P 11 P 10 0 threonine synthase Metabolism of small molecules b0008 talB 20561 P 20 P 13 P 16 P 13 0 transaldolase B Metabolism of small molecules b0009 mog 1296 P 7 0 0 0 0 required for the efficient incorporation of molybdate into molybdoproteins Metabolism of small molecules b0014 dnaK 13283 P 32 P 23 P 24 P 23 0 chaperone Hsp70; DNA biosynthesis; autoregulated heat shock proteins Cell processes b0031 dapB 2348 P 16 P 3 3 P 3 0 dihydrodipicolinate reductase Metabolism of small molecules b0032 carA 9312 P 14 P 8 P 8 P 8 0 carbamoyl-phosphate synthetase, glutamine (small) subunit Metabolism of small molecules b0048 folA 1588 P 7 P 1 2 P 1 0 dihydrofolate reductase type I; trimethoprim resistance Metabolism of small molecules peptidyl-prolyl cis-trans isomerase (PPIase), involved in maturation of outer b0053 surA 3825 P 19 P 4 P 5 P 4 P(m) 1 GenProt membrane proteins (1st module) Cell processes b0054 imp 2737 P 42 P 5 0 0 P(m) 5 GenProt organic solvent tolerance Cell processes b0071 leuD 4770 -

Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_001591825Cyc: Bacillus vietnamensis NBRC 101237 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari sn-glycerol phosphate phosphate pro phosphate phosphate phosphate thiamine molybdate D-xylose D-ribose glutathione 3-phosphate D-mannitol L-cystine L-djenkolate lanthionine α,β-trehalose phosphate phosphate [+ 3 more] α,α-trehalose predicted predicted ABC ABC FliY ThiT XylF RbsB RS10935 UgpC TreP PutP RS10200 PstB PstB RS10385 RS03335 RS20030 RS19075 transporter transporter of molybdate of phosphate α,β-trehalose 6-phosphate L-cystine D-xylose D-ribose sn-glycerol D-mannitol phosphate phosphate thiamine glutathione α α phosphate phosphate phosphate phosphate L-djenkolate 3-phosphate , -trehalose 6-phosphate pro 1-phosphate lanthionine molybdate phosphate [+ 3 more] Metabolic Regulator Amino Acid Degradation Amine and Polyamine Biosynthesis Macromolecule Modification tRNA-uridine 2-thiolation Degradation ATP biosynthesis a mature peptidoglycan a nascent β an N-terminal- -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

ADSL, AMPD1, and ATIC Expression Levels in Muscle and Their Correlations with Muscle Inosine Monophosphate Content in Dapulian and Hybridized Pig Species
Open Journal of Animal Sciences, 2017, 7, 393-404 http://www.scirp.org/journal/ojas ISSN Online: 2161-7627 ISSN Print: 2161-7597 ADSL, AMPD1, and ATIC Expression Levels in Muscle and Their Correlations with Muscle Inosine Monophosphate Content in Dapulian and Hybridized Pig Species Rongsheng Zhu1,2, Yanping Wang1,2, Huaizhong Wang1,2, Song Lin1,2, Shouli Sun1,2, Baohua Huang1,2, Hongmei Hu1,2* 1Institute of Animal Science and Veterinary Medicine, Shandong Academy of Agricultural Sciences, Jinan, China 2Shandong Key Laboratory of Animal Disease Control and Breeding, Jinan, China How to cite this paper: Zhu, R.S., Wang, Abstract Y.P., Wang, H.Z., Lin, S., Sun, S.L., Huang, B.H. and Hu, H.M. (2017) ADSL, AMPD1, We investigated the relationship between muscle inosine monophosphate and ATIC Expression Levels in Muscle and (IMP) content and mRNA levels of ADSL, AMPD1, and ATIC in Dapulian Their Correlations with Muscle Inosine (DPL), Landrace × Dapulian (LDPL), and Duroc × Landrace × Dapulian Monophosphate Content in Dapulian and Hybridized Pig Species. Open Journal of (DLDPL) hybridized pigs. Methods: The total RNA in longissimus dorsi was Animal Sciences, 7, 393-404. isolated from Dapulian (DPL), Landrace × Dapulian (LDPL) and Duroc × https://doi.org/10.4236/ojas.2017.74030 Landrace × Dapulian (DLDPL) hybridized pigs, weighed about 95.0 kg, n = 8/species. The internal genes with highest stability (YWHAZ and RPL4) were Received: July 16, 2017 Accepted: September 11, 2017 chosen from 11 common internal genes using Quantitative real-time PCR Published: September 14, 2017 (qPCR) and geNorm software. The mRNA levels of ADSL, AMPD1 and ATIC genes were corrected with YWHAZ and RPL4 genes. -

Metabolic Enzyme Expression Highlights a Key Role for MTHFD2 and the Mitochondrial Folate Pathway in Cancer
ARTICLE Received 1 Nov 2013 | Accepted 17 Dec 2013 | Published 23 Jan 2014 DOI: 10.1038/ncomms4128 Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer Roland Nilsson1,2,*, Mohit Jain3,4,5,6,*,w, Nikhil Madhusudhan3,4,5, Nina Gustafsson Sheppard1,2, Laura Strittmatter3,4,5, Caroline Kampf7, Jenny Huang8, Anna Asplund7 & Vamsi K. Mootha3,4,5 Metabolic remodeling is now widely regarded as a hallmark of cancer, but it is not clear whether individual metabolic strategies are frequently exploited by many tumours. Here we compare messenger RNA profiles of 1,454 metabolic enzymes across 1,981 tumours spanning 19 cancer types to identify enzymes that are consistently differentially expressed. Our meta- analysis recovers established targets of some of the most widely used chemotherapeutics, including dihydrofolate reductase, thymidylate synthase and ribonucleotide reductase, while also spotlighting new enzymes, such as the mitochondrial proline biosynthetic enzyme PYCR1. The highest scoring pathway is mitochondrial one-carbon metabolism and is centred on MTHFD2. MTHFD2 RNA and protein are markedly elevated in many cancers and correlated with poor survival in breast cancer. MTHFD2 is expressed in the developing embryo, but is absent in most healthy adult tissues, even those that are proliferating. Our study highlights the importance of mitochondrial compartmentalization of one-carbon metabolism in cancer and raises important therapeutic hypotheses. 1 Unit of Computational Medicine, Department of Medicine, Karolinska Institutet, 17176 Stockholm, Sweden. 2 Center for Molecular Medicine, Karolinska Institutet, 17176 Stockholm, Sweden. 3 Broad Institute, Cambridge, Massachusetts 02142, USA. 4 Department of Systems Biology, Harvard Medical School, Boston, Massachusetts 02115, USA. -

Surveying Purine Biosynthesis Across the Domains of Life Unveils
1 2 3 4 Article type : Special Feature Review 5 6 7 8 9 Surveying purine biosynthesis across the domains of life 10 unveils promising drug targets in pathogens 11 12 13 14 Sheena M. H. Chua and James A. Fraser* 15 16 17 18 Australian Infectious Diseases Research Centre 19 School of Chemistry & Molecular Biosciences 20 The University of Queensland, Brisbane, Queensland, Australia 21 22 23 24 25 26 27 28 29 30 Manuscript Author 31 *Correspondence e-mail: [email protected] This is the author manuscript accepted for publication and has undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/IMCB.12389 This article is protected by copyright. All rights reserved 32 Running head 33 34 Purine biosynthesis across the domains of life 35 36 37 Keywords 38 39 Drug target, gene fusions, immunotherapy, infectious diseases, purine metabolism, 40 purinosome 41 42 Abstract 43 Purines play an integral role in cellular processes such as energy metabolism, cell signalling, 44 and encoding the genetic makeup of all living organisms, ensuring that the purine metabolic 45 pathway is maintained across all domains of life. To gain a deeper understanding of purine 46 biosynthesis via the de novo biosynthetic pathway, the genes encoding purine metabolic 47 enzymes from 35 archaea, 69 bacteria and 99 eukaryotic species were investigated. While the 48 classic elements of the canonical purine metabolic pathway were utilised in all domains, a 49 subset of familiar biochemical roles were found to be performed by unrelated proteins in 50 some members of the Archaea and Bacteria. -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc. -
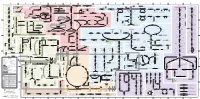
Fullsubwaymap221.Pdf
A B C D E F G H I J K L M Aldose reductase Sorbitol dehydrogenase Cholesterol synthesis Glycogen and galactose metabolism (branch point only) (debranching) glucose sorbitol fructose Aromatic amino acid metabolism Purine synthesis Pyrimidine synthesis glycogen (n+1) phenylacetate triiodothyronine (T3) dopaquinone melanin + + thyroxine (T4) (multiple steps) (many cycles) P 4' NADPH NADP NAD NADH acetyl-CoA x2 i Debranching enzyme 3' - α-1,4 bonds Aromatic amino acid 5-phosphoribosyl pyrophosphate (PRPP) HCO B 2' P 3 (branching) Glycogen 6 i (multiple steps) O H O (multiple steps) Tyrosinase O H O 1' 2 2 2 2 B 1 2 3 4 5 6 7 8 9 10 Pentose phosphate pathway Phenylalanine Tyrosine decarboxylase 6 Dopamine B Thiolase phosphorylase -5 -4 -3 -2 -1 0 1 2 3 4 Transaminase 6 glutamine 2 ATP (many cycles) Glycogen Glucose-6-phosphatase Dehydrogenase hydroxylase hydroxylase (DOPA decarboxylase) β-hydroxylase Methyltransferase 10 UDP 4:4 Transferase ATP glutathione (reduced) H O phenyllactate phenylpyruvate phenylalanine tyrosine dihydroxy- dopamine Glutamine PRPP CoA synthase Hexokinase or (in endoplasmic reticulum) 2 2 norepinephrine epinephrine glutamine Carbamoyl 9 1' phenylanine amidotransferase (GPAT) glutamate glycogen (n) Glutathione reductase Glutathione peroxidase + phosphate glycogen (n) -5 -4 -3 -2 -1 0 1 2 3 4 2' 3' 4' glucokinase 8 NADP NADPH glutamate α-ketoglutarate CO2 O H O Glycosyl (4:6) UDP-glucose ADP (L-DOPA) 2 2 synthetase II glutathione (oxidized) 2 H O α-ketoglutarate PPi glutamate acetoacetyl-CoA 7 1 Glucose -1,6-Glucosidase -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2. -

Protein T1 C1 Accession No. Description
Protein T1 C1 Accession No. Description SW:143B_HUMAN + + P31946 14-3-3 protein beta/alpha (protein kinase c inhibitor protein-1) (kcip-1) (protein 1054). 14-3-3 protein epsilon (mitochondrial import stimulation factor l subunit) (protein SW:143E_HUMAN + + P42655 P29360 Q63631 kinase c inhibitor protein-1) (kcip-1) (14-3-3e). SW:143S_HUMAN + - P31947 14-3-3 protein sigma (stratifin) (epithelial cell marker protein 1). SW:143T_HUMAN + - P27348 14-3-3 protein tau (14-3-3 protein theta) (14-3-3 protein t-cell) (hs1 protein). 14-3-3 protein zeta/delta (protein kinase c inhibitor protein-1) (kcip-1) (factor SW:143Z_HUMAN + + P29312 P29213 activating exoenzyme s) (fas). P01889 Q29638 Q29681 Q29854 Q29861 Q31613 hla class i histocompatibility antigen, b-7 alpha chain precursor (mhc class i antigen SW:1B07_HUMAN + - Q9GIX1 Q9TP95 b*7). hla class i histocompatibility antigen, b-14 alpha chain precursor (mhc class i antigen SW:1B14_HUMAN + - P30462 O02862 P30463 b*14). P30479 O19595 Q29848 hla class i histocompatibility antigen, b-41 alpha chain precursor (mhc class i antigen SW:1B41_HUMAN + - Q9MY79 Q9MY94 b*41) (bw-41). hla class i histocompatibility antigen, b-42 alpha chain precursor (mhc class i antigen SW:1B42_HUMAN + - P30480 P79555 b*42). P30488 O19615 O19624 O19641 O19783 O46702 hla class i histocompatibility antigen, b-50 alpha chain precursor (mhc class i antigen SW:1B50_HUMAN + - O78172 Q9TQG1 b*50) (bw-50) (b-21). hla class i histocompatibility antigen, b-54 alpha chain precursor (mhc class i antigen SW:1B54_HUMAN + - P30492 Q9TPQ9 b*54) (bw-54) (bw-22). P30495 O19758 P30496 hla class i histocompatibility antigen, b-56 alpha chain precursor (mhc class i antigen SW:1B56_HUMAN - + P79490 Q9GIM3 Q9GJ17 b*56) (bw-56) (bw-22).