New Targets in Malaria Parasite Chemotherapy: a Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Neena Valecha1, Deepali Savargaonkar1, Bina Srivastava1, B
Valecha et al. Malar J (2016) 15:42 DOI 10.1186/s12936-016-1084-1 Malaria Journal RESEARCH Open Access Comparison of the safety and efficacy of fixed‑dose combination of arterolane maleate and piperaquine phosphate with chloroquine in acute, uncomplicated Plasmodium vivax malaria: a phase III, multicentric, open‑label study Neena Valecha1, Deepali Savargaonkar1, Bina Srivastava1, B. H. Krishnamoorthy Rao2, Santanu K. Tripathi3, Nithya Gogtay4, Sanjay Kumar Kochar5, Nalli Babu Vijaya Kumar6, Girish Chandra Rajadhyaksha7, Jitendra D. Lakhani8, Bhagirath B. Solanki9, Rajinder K. Jalali10, Sudershan Arora10, Arjun Roy10, Nilanjan Saha10, Sunil S. Iyer10, Pradeep Sharma10 and Anupkumar R. Anvikar1* Abstract Background: Chloroquine has been the treatment of choice for acute vivax malaria for more than 60 years. Malaria caused by Plasmodium vivax has recently shown resistance to chloroquine in some places. This study compared the efficacy and safety of fixed dose combination (FDC) of arterolane maleate and piperaquine phosphate (PQP) with chloroquine in the treatment of uncomplicated vivax malaria. Methods: Patients aged 13–65 years with confirmed mono-infection of P. vivax along with fever or fever in the previ- ous 48 h were included. The 317 eligible patients were randomly assigned to receive FDC of arterolane maleate and PQP (n 159) or chloroquine (n 158) for 3 days. Primaquine was given as an anti-relapse measure on day 3 and continued= for 14 consecutive days.= Primary efficacy analysis included assessment of the proportion of aparasitaemic and afebrile patients at 72 h. Safety endpoints were analysis of adverse events, vital signs, laboratory data, and abnor- malities on electrocardiograph. Patients participated in the study for at least 42 days. -

Review Article Efforts Made to Eliminate Drug-Resistant Malaria and Its Challenges
Hindawi BioMed Research International Volume 2021, Article ID 5539544, 12 pages https://doi.org/10.1155/2021/5539544 Review Article Efforts Made to Eliminate Drug-Resistant Malaria and Its Challenges Wote Amelo 1,2,3 and Eyasu Makonnen 1,2 1Department of Pharmacology and Clinical Pharmacy, School of Pharmacy, Addis Ababa University, Addis Ababa, Ethiopia 2Center for Innovative Drug Development and Therapeutic Trials for Africa (CDT-Africa), Addis Ababa University, Addis Ababa, Ethiopia 3Department of Pharmacology and Toxicology, School of Pharmacy, Jimma University, Jimma, Ethiopia Correspondence should be addressed to Wote Amelo; [email protected] Received 21 January 2021; Accepted 9 August 2021; Published 30 August 2021 Academic Editor: Jane Hanrahan Copyright © 2021 Wote Amelo and Eyasu Makonnen. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Since 2000, a good deal of progress has been made in malaria control. However, there is still an unacceptably high burden of the disease and numerous challenges limiting advancement towards its elimination and ultimate eradication. Among the challenges is the antimalarial drug resistance, which has been documented for almost all antimalarial drugs in current use. As a result, the malaria research community is working on the modification of existing treatments as well as the discovery and development of new drugs to counter the resistance challenges. To this effect, many products are in the pipeline and expected to be marketed soon. In addition to drug and vaccine development, mass drug administration (MDA) is under scientific scrutiny as an important strategy for effective utilization of the developed products. -

Update Tot 30-04-2020 1. Chloroquine and Hydroxychloroquine for The
Update tot 30-04-2020 1. Chloroquine and Hydroxychloroquine for the Prevention or Treatment of Novel Coronavirus Disease (COVID-19) in Africa: Caution for Inappropriate Off-Label Use in Healthcare Settings. Abena PM, Decloedt EH, Bottieau E, Suleman F, Adejumo P, Sam-Agudu NA, et al. Am j trop med hyg. 2020. 2. Evaluation of Hydroxychloroquine Retinopathy Using Ultra-Widefield Fundus Autofluorescence: Peripheral Findings in the Retinopathy. Ahn SJ, Joung J, Lee BR. American journal of ophthalmology. 2020;209:35-44. http://dx.doi.org/10.1016/j.ajo.2019.09.008. Epub 2019 Sep 14. 3. COVID-19 research has overall low methodological quality thus far: case in point for chloroquine/hydroxychloroquine. Alexander PE, Debono VB, Mammen MJ, Iorio A, Aryal K, Deng D, et al. J clin epidemiol. 2020. 4. Chloroquine and Hydroxychloroquine in the Era of SARS - CoV2: Caution on Their Cardiac Toxicity. Bauman JL, Tisdale JE. Pharmacotherapy. 2020. 5. Repositioned chloroquine and hydroxychloroquine as antiviral prophylaxis for COVID-19: A protocol for rapid systematic review of randomized controlled trials. Chang R, Sun W-Z. medRxiv. 2020:2020.04.18.20071167. 6. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Colson P, Rolain J-M, Lagier J-C, Brouqui P, Raoult D. Int J Antimicrob Agents. 2020:105932-. 7. Dose selection of chloroquine phosphate for treatment of COVID-19 based on a physiologically based pharmacokinetic model. Cui C, Zhang M, Yao X, Tu S, Hou Z, Jie En VS, et al. Acta Pharmaceutica Sinica B. 2020. 8. Hydroxychloroquine; Why It Might Be Successful and Why It Might Not Be Successful in the Treatment of Covid-19 Pneumonia? Could It Be A Prophylactic Drug? Deniz O. -
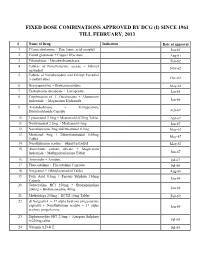
Fixed Dose Combinations Approved by Dcg (I) Since 1961 Till February, 2013
FIXED DOSE COMBINATIONS APPROVED BY DCG (I) SINCE 1961 TILL FEBRUARY, 2013 # Name of Drug Indication Date of approval 1. Cyanocobalamine + Zinc tannic acid complex Jan-61 2. Cobalt glutamate + Copper Glycinate Aug-61 3. Fibrinolysin + Desoxyribonuclease Feb-62 4. Tablets of Norethisterone acetate + Ethinyl Nov-62 oestradiol 5. Tablets of Norethynodrel and Ethinyl Estradiol 3-methyl ether Dec-62 6. Broxyquinoline + Brobenzoxalidine May-63 7. Testosterone decanoate + Isocaproate Jan-64 8. Combination of L Oxethazaine + Aluminium hydroxide + Magnesium Hydroxide Jun-66 9. Amylobarbitone + Trifluperazine Dihydrochloride Capsule Feb-67 10. Lynestronol 2.5mg + Mestranol 0.075mg Tablet Apr-67 11. Northynodrel 2.5mg + Mestranol 0.1mg Jun-67 12. Norethisterone 2mg and Mestranol 0.1mg May-67 13. Mestranol 4mg + Ethinyloestradial 0.05mg May-67 Tablet 14. Norethisterane acetate + ethinyl estradiol May-67 15. Aluminium sodium silicate + Magnesium hydroxide + Methypolysiloxane Tablet Jun-67 16. Ammoidin + Amidine Jul-67 17. Fluocortolene + Flucortolene Caproate Jul-68 18. Norgestrel + Ethinyloestradiol Tablet Aug-68 19. Folic Acid 0.5mg + Ferrous Sulphate 150mg Jan-69 Capsule 20. Tetracycline HCl 250mg + Broxyquinoline 200mg + Brobenzoxadine 40mg Jan-69 21. Methyldopa 250mg + HCTZ 15mg Tablet Feb-69 22. dl Norgestrel + 17 alpha hydroxy progesterone caproate + Norethisterone acetate + 17 alpha Jan-69 acetoxy progesterone 23. Diphenoxylate HCl 2.5mg + Atropine Sulphate 0.025mg tablet Jul-69 24. Vitamin A,D & E Jul-69 25. Lutin 0.1gm + Vit 0.1gm + Vit K1 2.5mg + Dicalcium Phosphate 0.1gm + Carlozochrome Jul-69 Salicylate 1mg tablet 26. Vit K 1 5mg + Calcium Lactolionate 100 m g+ Carlozocrome Salicylate 2.5mg + Phenol 0.5% Jul-69 + Lignocaine Hcl 1% injection 27. -

PHARMACOLOGY of NEWER ANTIMALARIAL DRUGS: REVIEW ARTICLE Bhuvaneshwari1, Souri S
REVIEW ARTICLE PHARMACOLOGY OF NEWER ANTIMALARIAL DRUGS: REVIEW ARTICLE Bhuvaneshwari1, Souri S. Kondaveti2 HOW TO CITE THIS ARTICLE: Bhuvaneshwari, Souri S. Kondaveti. ‖Pharmacology of Newer Antimalarial Drugs: Review Article‖. Journal of Evidence based Medicine and Healthcare; Volume 2, Issue 4, January 26, 2015; Page: 431-439. ABSTRACT: Malaria is currently is a major health problem, which has been attributed to wide spread resistance of the anopheles mosquito to the economical insecticides and increasing prevalence of drug resistance to plasmodium falciparum. Newer drugs are needed as there is a continual threat of emergence of resistance to both artemisins and the partner medicines. Newer artemisinin compounds like Artemisone, Artemisnic acid, Sodium artelinate, Arteflene, Synthetic peroxides like arterolane which is a synthetic trioxolane cognener of artemisins, OZ439 a second generation synthetic peroxide are under studies. Newer artemisinin combinations include Arterolane(150mg) + Piperaquine (750mg), DHA (120mg) + Piperaquine(960mg) (1:8), Artesunate + Pyronardine (1:3), Artesunate + Chlorproguanil + Dapsone, Artemisinin (125mg) + Napthoquine (50mg) single dose and Artesunate + Ferroquine.Newer drugs under development including Transmission blocking compounds like Bulaquine, Etaquine, Tafenoquine, which are primaquine congeners, Spiroindalone, Trioxaquine DU 1302, Epoxamicin, Quinolone 3 Di aryl ether. Newer drugs targeting blood & liver stages which include Ferroquine, Albitiazolium – (SAR – 97276). Older drugs with new use in malaria like beta blockers, calcium channel blockers, protease inhibitors, Dihydroorotate dehydrogenase inhibitors, methotrexate, Sevuparin sodium, auranofin, are under preclinical studies which also target blood and liver stages. Antibiotics like Fosmidomycin and Azithromycin in combination with Artesunate, Chloroquine, Clindamycin are also undergoing trials for treatment of malaria. Vaccines - RTS, S– the most effective malarial vaccine tested to date. -

Drug Targets of the Heartworm, Dirofilaria Immitis
Drug Targets of the Heartworm, Dirofilaria immitis Inauguraldissertation zur Erlangung des Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Christelle Godel aus La Sagne (NE) und Domdidier (FR) Schweiz Avenches, 2012 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät auf Antrag von Prof. Dr. Jürg Utzinger Prof. Dr. Pascal Mäser P.D. Dr. Ronald Kaminsky Prof. Dr. Georg von Samson-Himmelstjerna Basel, den 26th of June 2012 Prof. Dr. M. Spiess Dekan To my husband and my daughter With all my love. Table of Content P a g e | 2 Table of Content Table of Content P a g e | 3 Acknowledgements ............................................................................................................... 5 Summary ............................................................................................................................... 8 Introduction ..........................................................................................................................11 Dirofilaria immitis ..............................................................................................................12 Phylogeny and morphology ...........................................................................................12 Repartition and ecology .................................................................................................14 Life cycle .......................................................................................................................16 -

Synthetic Ozonide Drug Candidate OZ439 Offers New Hope for a Single-Dose Cure of Uncomplicated Malaria
Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria Susan A. Charmana, Sarah Arbe-Barnesb, Ian C. Bathurstc, Reto Brund,e, Michael Campbella, William N. Charmana, Francis C. K. Chiua, Jacques Cholletd,e, J. Carl Craftc, Darren J. Creeka, Yuxiang Dongf, Hugues Matileg, Melanie Maurerd,e, Julia Morizzia, Tien Nguyena, Petros Papastogiannidisd,e, Christian Scheurerd,e, David M. Shackleforda, Kamaraj Sriraghavanf, Lukas Stingelina, Yuanqing Tangf, Heinrich Urwylerh, Xiaofang Wangf, Karen L. Whitea, Sergio Wittlind,e, Lin Zhouf, and Jonathan L. Vennerstromf,1 aCentre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia; bFulcrum Pharma Developments Ltd., Hemel Hempstead, Hertfordshire HP1 1JY, United Kingdom; cMedicines for Malaria Venture, CH-1215 Geneva, Switzerland; dSwiss Tropical and Public Health Institute, CH-4002 Basel, Switzerland; eUniversity of Basel, CH-4051 Basel, Switzerland; fCollege of Pharmacy, University of Nebraska Medical Center, Omaha, NE 68198-6025; gF. Hoffmann-La Roche Ltd., CH-4070 Basel, Switzerland; and hBasilea Pharmaceutica Ltd., CH-4058 Basel, Switzerland Edited by Thomas E. Wellems, National Institutes of Health, Bethesda, MD, and approved January 11, 2011 (received for review October 21, 2010) Ozonide OZ439 is a synthetic peroxide antimalarial drug candidate Thai-Cambodian border, and more recently, increased parasite designed to provide a single-dose oral cure in humans. OZ439 has clearance times with artesunate (AS) monotherapy, have raised successfully completed Phase I clinical trials, where it was shown to significant concerns that resistance to these agents may be be safe at doses up to 1,600 mg and is currently undergoing Phase emerging (2, 3). -

A Variant Pfcrt Isoform Can Contribute to Plasmodium Falciparum Resistance to the First-Line Partner Drug Piperaquine" (2017)
Old Dominion University ODU Digital Commons Biological Sciences Faculty Publications Biological Sciences 5-2017 A Variant PfCRT Isoform Can Contribute to Plasmodium Falciparum Resistance to the First- Line Partner Drug Piperaquine Satish K. Dhingra Devasha Redhi Jill M. Combrinck Tomas Yeo John Okombo See next page for additional authors Follow this and additional works at: https://digitalcommons.odu.edu/biology_fac_pubs Part of the Biology Commons, Parasitic Diseases Commons, and the Pharmacology Commons Repository Citation Dhingra, Satish K.; Redhi, Devasha; Combrinck, Jill M.; Yeo, Tomas; Okombo, John; Henrich, Philipp P.; Cowell, Annie N.; Gupta, Purva; Stegman, Matthew L.; Hoke, Jonathan M.; Cooper, Roland A.; Winzeler, Elizabeth; Mok, Sachel; Egan, Timothy J.; and Fidock, David A., "A Variant PfCRT Isoform Can Contribute to Plasmodium Falciparum Resistance to the First-Line Partner Drug Piperaquine" (2017). Biological Sciences Faculty Publications. 211. https://digitalcommons.odu.edu/biology_fac_pubs/211 Original Publication Citation Dhingra, S. K., Redhi, D., Combrinck, J. M., Yeo, T., Okombo, J., Henrich, P. P., . Fidock, D. A. (2017). A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. MBio, 8(3). e00303-17 doi:10.1128/ mBio.00303-17 Authors Satish K. Dhingra, Devasha Redhi, Jill M. Combrinck, Tomas Yeo, John Okombo, Philipp P. Henrich, Annie N. Cowell, Purva Gupta, Matthew L. Stegman, Jonathan M. Hoke, Roland A. Cooper, Elizabeth Winzeler, Sachel Mok, Timothy J. Egan, and David A. Fidock This article is available at ODU Digital Commons: https://digitalcommons.odu.edu/biology_fac_pubs/211 Downloaded from RESEARCH ARTICLE crossm mbio.asm.org A Variant PfCRT Isoform Can Contribute on May 16, 2017 - Published by to Plasmodium falciparum Resistance to the First-Line Partner Drug Piperaquine Satish K. -

Dondorp, Background on Malaria and Combination Anti-Malarial Drug Therapy
Background on Malaria and Combination Anti-Malarial Drug Therapy FDA Workshop: Clinical Trial Design Considerations for Malaria Drug Development 30 June 2016 Prof. Arjen M. Dondorp Mahidol Oxford Tropical Medicine Research Unit The 5 human Plasmodium species P. falciparum P. vivax Anopheles P. knowlesi P. malariae P. ovale H. sapiens M. fascicularis/ nemestrina Life-cycle of Plasmodium White et al; Lancet 2014 Artemisinins: the best drugs for reducing malaria mortality Log-rank p=0.0002 SEAQUAMAT Asia 4 countries N=1,461 Δ=35% (202 children) AQUAMAT Artesunate Africa 9 countries Δ=23% N=5,425 Quinine (all children) Dondorp et al. Lancet 2010; SEAQUAMAT investigators group. Lancet 2005 Broader stage specificity explains superiority of artemisins rapid action, broad stage specificity, safe, easy administration White; Science 2009 Differences in potency TOTAL PARASITES Artesunate best drug to treat severe malaria 1012 Fastest for the artemisinins 1010 Detection limit 108 106 104 104 103 102 10 Reduction/ 48h-cycle 102 MQ, PQP, 0 Artesunate Malarone Doxycycline 0 1 2 3 4 WEEKS = artemisinin resistance Courtesy NJ White Differences in pharmacokinetics 100% M C P Plasma concentration (%) concentration Plasma A Q 0% 0 1 2 3 4 Time after start treatment (weeks) Courtesy NJ White Artemisinin resistance: a prelude to ACT failure 1. W-Cambodia 2007-2008 2012-2013 2012-2014 Slow clearance DHA-piperquine efficacy DHA-piperquine 42-day failures Source CNM Cambodia/ WHO Map by Richard Maude Dondorp et al. Amaratunga et al. N Eng J Med 2009 Lancet Infect Dis 2016 The molecular marker for artemisinin resistance: Kelch 13 K13 mutations in the “propeller region” strongly associates with the slow clearing phenotype multiple SNPs in the propeller region, but: only 1 mutation per clone seems permitted Ariey et al. -

ESCMID Online Lecture Library @ by Author Use of Antimalarials
Antimalarials for eradication ESCMID Online Lecture Library @ by author Use of antimalarials • Therapy • Prophylaxis • Intermittent preventive therapy in pregnancy • Intermittent preventive therapy in infants • Seasonal intermittent preventive therapy • Mass drug administration AndESCMID something Onlineelse ... Lecture Library @ by author The ideal antimalarial for eradication • Safe • Well tolerated • Efficacious and effective • No population restriction • Cheap • Active against all Plasmodium species • Active against all stages • Active against hypnozoites ESCMID Online Lecture Library @ by author The ideal antimalarial for eradication • One dose • Oral administration • No resistant parasites present • Refractory to resistance development • Fast acting • Long half life • Matching pharmacokinetics ESCMID Online Lecture Library @ by author Single Encounter Radical Cure and Prophylaxis Eliminate the human reservoir of infection Single patient encounter Radical cure leading to elimination of persistent asexual blood-stage forms and hypnozoites Prophylaxis to prevent reinfection for at least 1 month Suitable for mass drug administration (PLOS Medicine 2011) ESCMID Online Lecture Library @ by author ? Which drugs are there ESCMID Online Lecture Library @ by author List of antimalarials (1) Quinolines and combination of quinolines • quinine • quinine-clindamycin • quinine-doxycycline • quinine-tetracycline • quinine-sulfadoxine-pyrimethamine • quinidine • chloroquine • chloroquine-sulfadoxine-pyrimethamine • ESCMIDchloroquine-azithromycin -

Artemisinin and Quinoline Hybrid Compounds Inhibit Replication of SARS-Cov-2 in Vitro
Artemisinin and Quinoline Hybrid Compounds Inhibit Replication of SARS-CoV-2 In Vitro Lars Herrmann,[a] Ivan A. Yaremenko,[b] Aysun Çapcı,[a] Julia Struwe,[c] Jan Hodek,[d] Yulia Yu. Belyakova,[b] Peter S. Radulov,[b] Grigoriy A. Stepanov,[e] Jan Weber,[d] Alexander O. Terent'ev,*[b] Lutz Ackermann,*[c,f] and Svetlana B. Tsogoeva*[a] [a] L. Herrmann, Dr. A. Çapcı, Prof. Dr. S. B. Tsogoeva Organic Chemistry Chair I and Interdisciplinary Center for Molecular Materials (ICMM) Friedrich-Alexander University of Erlangen-Nürnberg Nikolaus Fiebiger-Straße 10, 91058 Erlangen, Germany E-mail: [email protected] [b] Dr. I. A. Yaremenko, Yu. Yu. Belyakova, Dr. P.S. Radulov, Prof. Dr. A.O. Terent’ev N. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences 47 Leninsky prosp., 119991 Moscow, Russian Federation. E-Mail: [email protected] [c] J. Struwe, Prof. Dr. L. Ackermann Institut für Organische und Biomolekulare Chemie Georg-August-Universität Göttingen Tammannstraße 2, 37077 Göttingen, Germany E-mail: [email protected] [d] Dr. J. Hodek, Dr. J. Weber Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences Flemingovo namesti 2, 16610 Prague, Czech Republic [e] Grigoriy A. Stepanov National Research University Higher School of Economics, Moscow, Russian Federation [f] Prof. Dr. L. Ackermann German Center for Cardiovascular Research (DZHK), Germany Abstract: The newly emerged severe acute respiratory syndrome by the World Health Organization (WHO) on March 11th 2020.[1] coronavirus 2 (SARS-CoV-2) cause life-threatening diseases in Several types of vaccines have already been developed, whereas millions of people worldwide and there is an urgent need for antiviral effective antiviral treatments are lacking. -

Late Breaker Abstracts
Late Breaker Abstracts 2480 CLONING AND CHARACTERIZATION OF SCHISTOSOMA JAPONICUM INSULIN RECEPTORS: POTENTIAL NEW INTERVENTION TARGETS AGAINST SCHISTOSOMIASIS Hong You, Wenbao Zhang, Malcolm K. Jones, Geoffrey N. Gobert, Donald P. McManus Queensland Institute of Medical Research, Brisbane, Australia Adult schistosomes depend for growth and development on hormonal signals from the mammalian host, which may include the insulin signalling pathway. To determine the precise role of insulin receptors in schistosome biology, we isolated two types of insulin receptors from Schistosoma japonicum, S. japonicum insulin receptor 1 (SjIR1) and SjIR2, with features similar to insulin receptors from other taxa. The sequences share 70% and 74% sequence identity to S. mansoni insulin receptor 1 and 2 (SmIR1 and SmIR2), respectively. SjIR1 and SjIR2 are conserved in tyrosine kinase domain to the other IRs, such as humans, mouse and Drosophila melanogaster. Phylogenetic analysis showed that SjIR2 and SmIR2 are close to Echinococcus multilocularis insulin receptor (EmIR), which is only one insulin receptor isolated in the tapeworm, indicating that SjIR2, SmIR2 and EmIR may be orthologs sharing the similar roles in the both schistosomes and Echinococcus, while IR1 homolog in E. multilocularis would have been lost during the cestode evolution. Real time PCR showed that the SjIRs were differentially expressed in different stages of S. japonisum, mainly in the stages in mammalian host, suggesting SjIRs may be involved in the host-parasite crosstalk. Yeast two-hybrid