Investigating the Mechanism of Action of Artemisinin Antimalarials and the Role of Ferriprotoporphyrin Ix Heme
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Role of Histidine-Rich Proteins in the Biomineralization of Hemozoin Lisa Pasierb
Duquesne University Duquesne Scholarship Collection Electronic Theses and Dissertations Fall 2005 The Role of Histidine-Rich Proteins in the Biomineralization of Hemozoin Lisa Pasierb Follow this and additional works at: https://dsc.duq.edu/etd Recommended Citation Pasierb, L. (2005). The Role of Histidine-Rich Proteins in the Biomineralization of Hemozoin (Doctoral dissertation, Duquesne University). Retrieved from https://dsc.duq.edu/etd/1021 This Immediate Access is brought to you for free and open access by Duquesne Scholarship Collection. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Duquesne Scholarship Collection. For more information, please contact [email protected]. The Role of Histidine-Rich Proteins in the Biomineralization of Hemozoin A Dissertation presented to the Bayer School of Natural and Environmental Sciences of Duquesne University As partial fulfillment of the requirements for the degree of Doctor of Philosophy By Lisa Pasierb August 26, 2005 Dr. David Seybert, thesis director Dr. David W. Wright, advisor In memory of Anna Pasierb April 24, 1924 – May 31, 2005 ii Acknowledgements First and foremost, I would like to express my sincerest gratitude to my advisor, Dr. David W. Wright. His exuberating energy and conviction attracted me to his research group, while his unwavering faith in me taught me more than he could ever know. Secondly, of course, I would like to extend my appreciation to Glenn Spreitzer and James Ziegler, the other two original members of the Wright group, whom initially tried to exert male dominance, but eventually became very faithful friends and colleagues. Finally, to all the other members of the Wright group over the years, thanks for all of your help, suggestions, and camaraderie. -
Neena Valecha1, Deepali Savargaonkar1, Bina Srivastava1, B
Valecha et al. Malar J (2016) 15:42 DOI 10.1186/s12936-016-1084-1 Malaria Journal RESEARCH Open Access Comparison of the safety and efficacy of fixed‑dose combination of arterolane maleate and piperaquine phosphate with chloroquine in acute, uncomplicated Plasmodium vivax malaria: a phase III, multicentric, open‑label study Neena Valecha1, Deepali Savargaonkar1, Bina Srivastava1, B. H. Krishnamoorthy Rao2, Santanu K. Tripathi3, Nithya Gogtay4, Sanjay Kumar Kochar5, Nalli Babu Vijaya Kumar6, Girish Chandra Rajadhyaksha7, Jitendra D. Lakhani8, Bhagirath B. Solanki9, Rajinder K. Jalali10, Sudershan Arora10, Arjun Roy10, Nilanjan Saha10, Sunil S. Iyer10, Pradeep Sharma10 and Anupkumar R. Anvikar1* Abstract Background: Chloroquine has been the treatment of choice for acute vivax malaria for more than 60 years. Malaria caused by Plasmodium vivax has recently shown resistance to chloroquine in some places. This study compared the efficacy and safety of fixed dose combination (FDC) of arterolane maleate and piperaquine phosphate (PQP) with chloroquine in the treatment of uncomplicated vivax malaria. Methods: Patients aged 13–65 years with confirmed mono-infection of P. vivax along with fever or fever in the previ- ous 48 h were included. The 317 eligible patients were randomly assigned to receive FDC of arterolane maleate and PQP (n 159) or chloroquine (n 158) for 3 days. Primaquine was given as an anti-relapse measure on day 3 and continued= for 14 consecutive days.= Primary efficacy analysis included assessment of the proportion of aparasitaemic and afebrile patients at 72 h. Safety endpoints were analysis of adverse events, vital signs, laboratory data, and abnor- malities on electrocardiograph. Patients participated in the study for at least 42 days. -

WO 2017/145013 Al 31 August 2017 (31.08.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/145013 Al 31 August 2017 (31.08.2017) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C07D 498/04 (2006.01) A61K 31/5365 (2006.01) kind of national protection available): AE, AG, AL, AM, C07D 519/00 (2006.01) A61P 25/00 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, (21) Number: International Application DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/IB20 17/050844 HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KH, KN, (22) International Filing Date: KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, 15 February 2017 (15.02.2017) MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, (25) Filing Language: English RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, (26) Publication Language: English TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 62/298,657 23 February 2016 (23.02.2016) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: PFIZER INC. [US/US]; 235 East 42nd Street, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, New York, New York 10017 (US). -

Chapter 5 High-Pressure Stopped-Flow Kinetic Studies of Nnos
Probing the dynamics and conformational landscape of neuronal nitric oxide synthase A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Life Sciences 2013 Anna Sobolewska-Stawiarz Contents Contents ...................................................................................................................... 2 List of Figures ............................................................................................................. 6 List of Tables ............................................................................................................ 10 Abstract ..................................................................................................................... 12 Declaration ................................................................................................................ 13 Copyright Statement ................................................................................................ 13 Acknowledgements ................................................................................................... 14 List of Abbreviations ............................................................................................... 15 List of Amino Acids Abbreviations ........................................................................ 17 CHAPTER 1. INTRODUCTION ........................................................................... 18 1.1 Nitric oxide synthase ......................................................................................... -

Dalbavancin • Oritavancin • Tedizolid • Ceftolozane/Tazobactam • Ceftazidime/Avibactam • Fecal Transplant
What’s New in Infectious Diseases? Bruce L. Gilliam, M.D. Institute of Human Virology University of Maryland School of Medicine Baltimore, MD Topics New Antibacterial Therapeutics Emerging Pathogens HIV Hepatitis C Disclosures Research Studies Pfizer – Staph aureus Vaccine Trial TaiMed Biologics - Ibaluzimab Advisory Board Viiv Healthcare New Antibacterial Therapeutics • Dalbavancin • Oritavancin • Tedizolid • Ceftolozane/tazobactam • Ceftazidime/avibactam • Fecal Transplant Incidence of Staph aureus hospitalizations in U.S.A., 2001–2009 BMC Infect Dis 2014, 14:296 Dalbavancin (Dalvance) • Derived from Teicoplanin • ½ life – Effective: 8.5 days – Terminal: 346 hrs (14 days) • Bactericidal • Similar spectrum to Vancomycin, active against: – Staphylococci • MSSA, MRSA, CoNS – Streptococci • resistant pneumococci • anaerobic strep – Enterococci • VRE with van B, C but not A – Corynebacterium Dalbavancin Once Weekly Non- Inferior to Vanco/Linezolid N Engl J Med 2014;370:2169-79. Single-Dose (1.5 g) Non-Inferior to Weekly Dalbavancin for Treatment of Acute Bacterial Skin and Skin Structure Infection 100 90 80 70 60 50 Single Dose 40 Once Weekly 30 20 10 0 Overall Clinical Success Rate Success Rate Success Rate Response Day 14 Day 28 Day 14 MRSA Clin Infect Dis. 2015 Nov 26. pii: civ982. [Epub ahead of print] VA Experience with Dalbavancin • Background – Levels in bone > MIC for 14 days • 8 patients treated for osteomyelitis with IV Dalbavancin – Former IV drug users not eligible for home IV or unwilling to do home IV • Treated for up to 8 weeks • No adverse events • All with resolution of osteomyelitis • Cost savings vs. placement in facility Oritavancin (Orbactiv) • Derived from Vancomycin • ½ life – Terminal 245-393 hrs (10-16 days) • Bactericidal • Similar spectrum to Vancomycin, active against: – Staphylococci • MSSA, MRSA, CoNS – Streptococci • resistant pneumococci • anaerobic strep – Enterococci • VRE with van A, B, C – Corynebacterium Single Dose Oritavancin vs. -

Review Article Efforts Made to Eliminate Drug-Resistant Malaria and Its Challenges
Hindawi BioMed Research International Volume 2021, Article ID 5539544, 12 pages https://doi.org/10.1155/2021/5539544 Review Article Efforts Made to Eliminate Drug-Resistant Malaria and Its Challenges Wote Amelo 1,2,3 and Eyasu Makonnen 1,2 1Department of Pharmacology and Clinical Pharmacy, School of Pharmacy, Addis Ababa University, Addis Ababa, Ethiopia 2Center for Innovative Drug Development and Therapeutic Trials for Africa (CDT-Africa), Addis Ababa University, Addis Ababa, Ethiopia 3Department of Pharmacology and Toxicology, School of Pharmacy, Jimma University, Jimma, Ethiopia Correspondence should be addressed to Wote Amelo; [email protected] Received 21 January 2021; Accepted 9 August 2021; Published 30 August 2021 Academic Editor: Jane Hanrahan Copyright © 2021 Wote Amelo and Eyasu Makonnen. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Since 2000, a good deal of progress has been made in malaria control. However, there is still an unacceptably high burden of the disease and numerous challenges limiting advancement towards its elimination and ultimate eradication. Among the challenges is the antimalarial drug resistance, which has been documented for almost all antimalarial drugs in current use. As a result, the malaria research community is working on the modification of existing treatments as well as the discovery and development of new drugs to counter the resistance challenges. To this effect, many products are in the pipeline and expected to be marketed soon. In addition to drug and vaccine development, mass drug administration (MDA) is under scientific scrutiny as an important strategy for effective utilization of the developed products. -

Jos Journal 2
POST-OPERATIVE AUDIT OF G6PD-DEFICIENT MALE CHILDREN WITH OBSTRUCTIVE ADENOTONSILLAR ENLARGEMENT AT UNIVERSITY COLLEGE HOSPITAL, IBADAN, NIGERIA. John EN1, Totyen EL1, Jacob N2, Nwaorgu OGB1 1 .Department of ENT/Head and Neck Surgery, University College Hospital, Ibadan, Nigeria 2. Department of paediatrics, University College Hospital, Ibadan, Nigeria All correspondences and request for reprint to Dr John EN, Department of ENT/Head and Neck Surgery, University College Hospital, Ibadan, Nigeria Email: [email protected] Telephone: +2348036240109 Abstract Background: G6PD deficiency ranks among the commonest hereditary enzyme deficiency worldwide and notable as a predisposing condition to haemolyticcrises. The fear of possible untoward effects is often expressed by parents of G6PD deficient male children scheduled for surgery after obtaining an informed and understood consent. The parental perception of obstructive adenotonsillar enlargement in this condition was also appraised. Methods: A retrospective chart review of all G6PD deficient male children between ages 1 to 7years who had adenotonsillectomy over a 3year period at University college Hospital, Ibadan, Nigeria. Results: The patients comprised of 22 G6PD deficient male children diagnosed shortly after birth upon development of neonatal jaundice. Fifteen(68.2%) and 6(27.3%) of the patients subsequently developed episodes of drug- induced haemolysis and non-haemolytic drug reactions prior to undergoing adenotonsillectomy by the otolaryngologists. None of the patients was observed to develop haemolytic crises up to 2weeks post-adenotonsillectomy. From the parental perception and responses in the follow-up period,all 22(100%) patient had resolution of noisy breathing, 20(91%) had improvement of snoring and apnoeic spells. Only 15 (68%) were reported to stop mouth-breathing. -

Update Tot 30-04-2020 1. Chloroquine and Hydroxychloroquine for The
Update tot 30-04-2020 1. Chloroquine and Hydroxychloroquine for the Prevention or Treatment of Novel Coronavirus Disease (COVID-19) in Africa: Caution for Inappropriate Off-Label Use in Healthcare Settings. Abena PM, Decloedt EH, Bottieau E, Suleman F, Adejumo P, Sam-Agudu NA, et al. Am j trop med hyg. 2020. 2. Evaluation of Hydroxychloroquine Retinopathy Using Ultra-Widefield Fundus Autofluorescence: Peripheral Findings in the Retinopathy. Ahn SJ, Joung J, Lee BR. American journal of ophthalmology. 2020;209:35-44. http://dx.doi.org/10.1016/j.ajo.2019.09.008. Epub 2019 Sep 14. 3. COVID-19 research has overall low methodological quality thus far: case in point for chloroquine/hydroxychloroquine. Alexander PE, Debono VB, Mammen MJ, Iorio A, Aryal K, Deng D, et al. J clin epidemiol. 2020. 4. Chloroquine and Hydroxychloroquine in the Era of SARS - CoV2: Caution on Their Cardiac Toxicity. Bauman JL, Tisdale JE. Pharmacotherapy. 2020. 5. Repositioned chloroquine and hydroxychloroquine as antiviral prophylaxis for COVID-19: A protocol for rapid systematic review of randomized controlled trials. Chang R, Sun W-Z. medRxiv. 2020:2020.04.18.20071167. 6. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Colson P, Rolain J-M, Lagier J-C, Brouqui P, Raoult D. Int J Antimicrob Agents. 2020:105932-. 7. Dose selection of chloroquine phosphate for treatment of COVID-19 based on a physiologically based pharmacokinetic model. Cui C, Zhang M, Yao X, Tu S, Hou Z, Jie En VS, et al. Acta Pharmaceutica Sinica B. 2020. 8. Hydroxychloroquine; Why It Might Be Successful and Why It Might Not Be Successful in the Treatment of Covid-19 Pneumonia? Could It Be A Prophylactic Drug? Deniz O. -

Annex 4: Drug Dosages for Children (Formulary)
Medicines Dosage Form Dose according to body weight (calculate if weight is below or over) 3-6 kg 6-10 kg 10-15 kg 15-20 kg 20-29 kg albendazole 200 mg (half tablet) 12-24 months chewable tablet, 400mg 400 mg (one tablet) over 24 months amodiaquine 10 mg base/kg/3 days (total dose 30 mg base/kg) tablet, 200mg - - 1 1 1 amoxicillin 15 mg/kg/dose for 7 days tablet/capsule 250 mg ¼ ½ ¾ 1 1½ oral suspension, 125mg/5ml 2.5 ml 5 ml 7.5 ml 10 ml - non-severe pneumonia: 25 mg/kg 2 times per day for 3 days tablet/capsule 250 mg ½ 1 1½ 2 2½ oral suspension, 125mg/5ml 5 ml 10 ml 15 ml - - ampicillin IM 50 mg/kg/6 hours Vial of 500 mg mixed with 2.1 ml 1 ml 2 ml 3 ml 5 ml 6 ml sterile water to give 500 mg/2.5 ml artemether IM 3.2 mg/kg once on day 1 injection, 40mg/ml in 1ml ampoule then injection, 80mg/ml in 1ml ampoule see Chapter 5, management of the child with malaria IM 1.6 mg/kg daily until oral therapy is possible, total therapy one week artemether + fixed dose treatment (20+120 mg) twice daily for 3 days tablet 10+120 mg see Chapter 5, management of the child with malaria lumefantrine artesunate severe malaria: IV or IM 2.4 mg/kg over 3 minutes at 0, 12 and 24 vial of 60 mg in 0.6 ml with 3.4 ml hours on day 1. -

Cytochrome P450 Oxidative Metabolism: Contributions to the Pharmacokinetics, Safety, and Efficacy of Xenobiotics
1521-009X/44/8/1229–1245$25.00 http://dx.doi.org/10.1124/dmd.116.071753 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 44:1229–1245, August 2016 Copyright ª 2016 by The American Society for Pharmacology and Experimental Therapeutics Special Section on Emerging Novel Enzyme Pathways in Drug Metabolism—Commentary Cytochrome P450 and Non–Cytochrome P450 Oxidative Metabolism: Contributions to the Pharmacokinetics, Safety, and Efficacy of Xenobiotics Robert S. Foti and Deepak K. Dalvie Pharmacokinetics and Drug Metabolism, Amgen, Cambridge, Massachusetts (R.S.F.); and Pharmacokinetics, Dynamics, and Metabolism, Pfizer, La Jolla, California (D.K.D.) Downloaded from Received May 24, 2016; accepted June 10, 2016 ABSTRACT The drug-metabolizing enzymes that contribute to the metabolism this end, this Special Section on Emerging Novel Enzyme Pathways or bioactivation of a drug play a crucial role in defining the in Drug Metabolism will highlight a number of advancements that dmd.aspetjournals.org absorption, distribution, metabolism, and excretion properties of have recently been reported. The included articles support the that drug. Although the overall effect of the cytochrome P450 (P450) important role of non-P450 enzymes in the clearance pathways of family of drug-metabolizing enzymes in this capacity cannot be U.S. Food and Drug Administration–approved drugs over the past understated, advancements in the field of non-P450–mediated me- 10 years. Specific examples will detail recent reports of aldehyde tabolism have garnered increasing attention in recent years. This is oxidase, flavin-containing monooxygenase, and other non-P450 perhaps a direct result of our ability to systematically avoid P450 pathways that contribute to the metabolic, pharmacokinetic, or liabilities by introducing chemical moieties that are not susceptible pharmacodynamic properties of xenobiotic compounds. -

CAS Number Index
2334 CAS Number Index CAS # Page Name CAS # Page Name CAS # Page Name 50-00-0 905 Formaldehyde 56-81-5 967 Glycerol 61-90-5 1135 Leucine 50-02-2 596 Dexamethasone 56-85-9 963 Glutamine 62-44-2 1640 Phenacetin 50-06-6 1654 Phenobarbital 57-00-1 514 Creatine 62-46-4 1166 α-Lipoic acid 50-11-3 1288 Metharbital 57-22-7 2229 Vincristine 62-53-3 131 Aniline 50-12-4 1245 Mephenytoin 57-24-9 1950 Strychnine 62-73-7 626 Dichlorvos 50-23-7 1017 Hydrocortisone 57-27-2 1428 Morphine 63-05-8 127 Androstenedione 50-24-8 1739 Prednisolone 57-41-0 1672 Phenytoin 63-25-2 335 Carbaryl 50-29-3 569 DDT 57-42-1 1239 Meperidine 63-75-2 142 Arecoline 50-33-9 1666 Phenylbutazone 57-43-2 108 Amobarbital 64-04-0 1648 Phenethylamine 50-34-0 1770 Propantheline bromide 57-44-3 191 Barbital 64-13-1 1308 p-Methoxyamphetamine 50-35-1 2054 Thalidomide 57-47-6 1683 Physostigmine 64-17-5 784 Ethanol 50-36-2 497 Cocaine 57-53-4 1249 Meprobamate 64-18-6 909 Formic acid 50-37-3 1197 Lysergic acid diethylamide 57-55-6 1782 Propylene glycol 64-77-7 2104 Tolbutamide 50-44-2 1253 6-Mercaptopurine 57-66-9 1751 Probenecid 64-86-8 506 Colchicine 50-47-5 589 Desipramine 57-74-9 398 Chlordane 65-23-6 1802 Pyridoxine 50-48-6 103 Amitriptyline 57-92-1 1947 Streptomycin 65-29-2 931 Gallamine 50-49-7 1053 Imipramine 57-94-3 2179 Tubocurarine chloride 65-45-2 1888 Salicylamide 50-52-2 2071 Thioridazine 57-96-5 1966 Sulfinpyrazone 65-49-6 98 p-Aminosalicylic acid 50-53-3 426 Chlorpromazine 58-00-4 138 Apomorphine 66-76-2 632 Dicumarol 50-55-5 1841 Reserpine 58-05-9 1136 Leucovorin 66-79-5 -
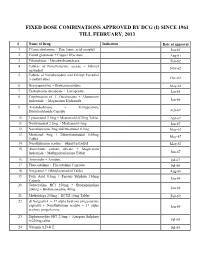
Fixed Dose Combinations Approved by Dcg (I) Since 1961 Till February, 2013
FIXED DOSE COMBINATIONS APPROVED BY DCG (I) SINCE 1961 TILL FEBRUARY, 2013 # Name of Drug Indication Date of approval 1. Cyanocobalamine + Zinc tannic acid complex Jan-61 2. Cobalt glutamate + Copper Glycinate Aug-61 3. Fibrinolysin + Desoxyribonuclease Feb-62 4. Tablets of Norethisterone acetate + Ethinyl Nov-62 oestradiol 5. Tablets of Norethynodrel and Ethinyl Estradiol 3-methyl ether Dec-62 6. Broxyquinoline + Brobenzoxalidine May-63 7. Testosterone decanoate + Isocaproate Jan-64 8. Combination of L Oxethazaine + Aluminium hydroxide + Magnesium Hydroxide Jun-66 9. Amylobarbitone + Trifluperazine Dihydrochloride Capsule Feb-67 10. Lynestronol 2.5mg + Mestranol 0.075mg Tablet Apr-67 11. Northynodrel 2.5mg + Mestranol 0.1mg Jun-67 12. Norethisterone 2mg and Mestranol 0.1mg May-67 13. Mestranol 4mg + Ethinyloestradial 0.05mg May-67 Tablet 14. Norethisterane acetate + ethinyl estradiol May-67 15. Aluminium sodium silicate + Magnesium hydroxide + Methypolysiloxane Tablet Jun-67 16. Ammoidin + Amidine Jul-67 17. Fluocortolene + Flucortolene Caproate Jul-68 18. Norgestrel + Ethinyloestradiol Tablet Aug-68 19. Folic Acid 0.5mg + Ferrous Sulphate 150mg Jan-69 Capsule 20. Tetracycline HCl 250mg + Broxyquinoline 200mg + Brobenzoxadine 40mg Jan-69 21. Methyldopa 250mg + HCTZ 15mg Tablet Feb-69 22. dl Norgestrel + 17 alpha hydroxy progesterone caproate + Norethisterone acetate + 17 alpha Jan-69 acetoxy progesterone 23. Diphenoxylate HCl 2.5mg + Atropine Sulphate 0.025mg tablet Jul-69 24. Vitamin A,D & E Jul-69 25. Lutin 0.1gm + Vit 0.1gm + Vit K1 2.5mg + Dicalcium Phosphate 0.1gm + Carlozochrome Jul-69 Salicylate 1mg tablet 26. Vit K 1 5mg + Calcium Lactolionate 100 m g+ Carlozocrome Salicylate 2.5mg + Phenol 0.5% Jul-69 + Lignocaine Hcl 1% injection 27.