NIH Public Access Author Manuscript Curr Opin Biotechnol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Purine and Pyrimidine Metabolism in Human Epidermis* Jean De Bersaques, Md
THE JOURNAL OP INVESTIGATIVE DERMATOLOGY Vol. 4s, No. Z Copyright 1957 by The Williams & Wilkins Co. Fri nte,1 in U.S.A. PURINE AND PYRIMIDINE METABOLISM IN HUMAN EPIDERMIS* JEAN DE BERSAQUES, MD. The continuous cellular renewal occurring inthine, which contained 5% impurity, and for uric the epidermis requires a very active synthesisacid, which consisted of 3 main components. The reaction was stopped after 1—2 hours in- and breakdown of nuclear and cytoplasmiecubation at 37° and the products were spotted on nucleic acids. Data on the enzyme systemsWhatman 1 filter paper sheets. According to the participating in these metabolic processes arereaction products expected, a choice was made of rather fragmentary (1—9) and some are, inat least 2 among the following solvents, all used terms of biochemical time, in need of up- in ascending direction: 1. isoamyl alcohol—5% Na2HPO4 (1:1), dating. In some other publications (10—18), 2. water-saturated n-butanol, the presence and concentration of various in- 3. distilled water, termediate products is given. 4. 80% formic acid—n-hutanol——n-propanol— In this paper, we tried to collect and supple- acetone—30% trichloro-aeetic acid (5:8:4: ment these data by investigating the presence 5:3), 5. n-butanol——4% boric acid (43:7), or absence in epidermis of enzyme systems 6. isobutyrie acid—water—ammonia 0.880—ver- that have been described in other tissues. sene 0.1M(500:279:21:8), This first investigation was a qualitative one, 7. upper phase of ethyl acetate—water—formic and some limitations were set by practical acid (12:7:1), 8. -

Deep Sequencing of the Camellia Sinensis Transcriptome Revealed
Shi et al. BMC Genomics 2011, 12:131 http://www.biomedcentral.com/1471-2164/12/131 RESEARCHARTICLE Open Access Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds Cheng-Ying Shi1†, Hua Yang1†, Chao-Ling Wei1†, Oliver Yu1,2, Zheng-Zhu Zhang1, Chang-Jun Jiang1, Jun Sun1, Ye-Yun Li1, Qi Chen1, Tao Xia1, Xiao-Chun Wan1* Abstract Background: Tea is one of the most popular non-alcoholic beverages worldwide. However, the tea plant, Camellia sinensis, is difficult to culture in vitro, to transform, and has a large genome, rendering little genomic information available. Recent advances in large-scale RNA sequencing (RNA-seq) provide a fast, cost-effective, and reliable approach to generate large expression datasets for functional genomic analysis, which is especially suitable for non-model species with un-sequenced genomes. Results: Using high-throughput Illumina RNA-seq, the transcriptome from poly (A)+ RNA of C. sinensis was analyzed at an unprecedented depth (2.59 gigabase pairs). Approximate 34.5 million reads were obtained, trimmed, and assembled into 127,094 unigenes, with an average length of 355 bp and an N50 of 506 bp, which consisted of 788 contig clusters and 126,306 singletons. This number of unigenes was 10-fold higher than existing C. sinensis sequences deposited in GenBank (as of August 2010). Sequence similarity analyses against six public databases (Uniprot, NR and COGs at NCBI, Pfam, InterPro and KEGG) found 55,088 unigenes that could be annotated with gene descriptions, conserved protein domains, or gene ontology terms. Some of the unigenes were assigned to putative metabolic pathways. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -
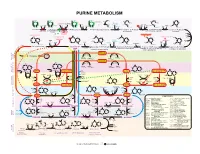
15 Purines Apr13.Ai
PURINE METABOLISM CH O-P CH O-P CH O-P CH O-P NH NH NH NH 2 2 2 2 H C 2 H C H C HC O H O H O NH O NHCOCH NH 2 2 2 2 2 2 CHO CHO CH Formyl- Glutamine Glutamate OC OC C C H H H H H H Glycine H H or NH H folate NH ADP N H OH H H H H 4 ATP Pi HN = NH ADP ATP A MP O-P-O-P H O H ATP ADP+P Gln H O Glu H N 2 PP H folate 2 ATP Pi 2 OH OH OH OH OH OH OH OH Ribose-P 4 Ribose-P Ribose-P Ribose-P 5-P-ribose 5-P-ribosyl- 5-P-Ribosylamine N1-(5-P-ribosyl)glycinamide N2-formyl-N1-(5-P-ribosyl) 2-(formamido)-N1- 5-amino-1-(P-ribosyl)- 2.7.6.1 pyrophosphate 2.4.2.14 6.3.4.13 2.1.2.2 6.3.5.3 (5-P-ribosyl)acetamidine 6.3.3.1 ADP GDP AMP GTP glycinamide - imidazole (PRPP) GDP ADP ATP ATPGMP -OOCCHCH COO- -OOCCHCH COO- 2 2 O O O CO NH - - - - 2 OOCCHCH COO C HN CO OOCCHCH2COO - C NH 2 C N C NH NH NH OOC 4.1.1.21 N 2 HN H N + NH N C C H2N C 2 C C Aspartate NH3 C CH GDP CH CH Formyl- CH CH Aspartate CH ADP HC C HC C OHC C H4folate C C Pi N Pi N H O N N N ADP C N GTP N 2 N H4folate H N H N N 2 Fumarate 2 Pi ATP H N Ribose-P Ribose-P H Ribose-P Ribose-P Ribose-P 2 E 4.3.2.2 Ribose-P YCL C Adenylosuccinate 4.3.2.2 E Inosine- 5-formamido-1-(5-P-ribosyl)- 5-amino-1-(5-P-ribosyl)- 5-amino-1-(5-P-ribosyl-) 5-amino-1-(5-P-ribosyl) ID P 3.5.4.10 2.1.2.3 T i m i d a z ole-4-(N)-[1,2-dicarboxy-6.3.2.6 O imidazole-4-carboxamide imidazole-4-carboxylate E IMP imidazole-4-carboxamide L ethyl]-carboxamide C U N C E I RNA N S E I 2.7.7.6 2.7.7.6 R D L TCA I Fumarate U C C CYCLE P U RNA A RNA RNA n N n n+1 RNAn+1 DNA S NH2 E 2.7.7.7 2.7.7.7 E NH C T 2 N O D A C I N N C H T N -

POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii
POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii http://rcin.org.pl WSKAZÓWKI DLA AUTORÓW Kwartalnik „Postępy Biochemii” publikuje artykuły monograficzne omawiające wąskie tematy, oraz artykuły przeglądowe referujące szersze zagadnienia z biochemii i nauk pokrewnych. Artykuły pierwszego typu winny w sposób syntetyczny omawiać wybrany temat na podstawie możliwie pełnego piśmiennictwa z kilku ostatnich lat, a artykuły drugiego typu na podstawie piśmiennictwa z ostatnich dwu lat. Objętość takich artykułów nie powinna przekraczać 25 stron maszynopisu (nie licząc ilustracji i piśmiennictwa). Kwartalnik publikuje także artykuły typu minireviews, do 10 stron maszynopisu, z dziedziny zainteresowań autora, opracowane na podstawie najnow szego piśmiennictwa, wystarczającego dla zilustrowania problemu. Ponadto kwartalnik publikuje krótkie noty, do 5 stron maszynopisu, informujące o nowych, interesujących osiągnięciach biochemii i nauk pokrewnych, oraz noty przybliżające historię badań w zakresie różnych dziedzin biochemii. Przekazanie artykułu do Redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii”. Autorzy artykułu odpowiadają za prawidłowość i ścisłość podanych informacji. Autorów obowiązuje korekta autorska. Koszty zmian tekstu w korekcie (poza poprawieniem błędów drukarskich) ponoszą autorzy. Artykuły honoruje się według obowiązujących stawek. Autorzy otrzymują bezpłatnie 25 odbitek swego artykułu; zamówienia na dodatkowe odbitki (płatne) należy zgłosić pisemnie odsyłając pracę po korekcie autorskiej. Redakcja prosi autorów o przestrzeganie następujących wskazówek: Forma maszynopisu: maszynopis pracy i wszelkie załączniki należy nadsyłać w dwu egzem plarzach. Maszynopis powinien być napisany jednostronnie, z podwójną interlinią, z marginesem ok. 4 cm po lewej i ok. 1 cm po prawej stronie; nie może zawierać więcej niż 60 znaków w jednym wierszu nie więcej niż 30 wierszy na stronie zgodnie z Normą Polską. -

Purine Metabolism
PURINE METABOLISM CH O-P CH O-P CH O-P CH O-P NH NH NH NH 2 2 2 2 H C 2 H C HC O H O H O O NHCOCH NH 2 2 H2C NH2 2 2 Formyl- CHO CHO CH Glutamine Glutamate OC OC C C H H H H H H Glycine H H or NH H folate NH ADP N H OH H H H H 4 ATP Pi HN = NH ADP ATP AMP O-P-O-P HO H ATP ADP+P Gln H O Glu H N 2 PP H folate 2 ATP Pi 2 OH OH OH OH OH OH OH OH Ribose-P 4 Ribose-P Ribose-P Ribose-P 5-P-ribose 5-P-ribosyl- 5-P-Ribosylamine N1-(5-P-ribosyl)glycinamide N2-formyl-N1-(5-P-ribosyl) 2-(formamido)-N1- 5-amino-1-(P-ribosyl)- 2.7.6.1 pyrophosphate 2.4.2.14 6.3.4.13 2.1.2.2 6.3.5.3 (5-P-ribosyl)acetamidine 6.3.3.1 ADP GDP AMP GTP glycinamide - imidazole (PRPP) GDP ADP ATP ATPGMP -OOCCHCH COO- -OOCCHCH COO- 2 2 O O O CO NH - - - - 2 OOCCHCH COO C HN CO OOCCHCH2COO - C NH 2 C N C NH NH NH OOC 4.1.1.21 N 2 HN H N + NH N C C H2N C 2 C C Aspartate NH3 C CH GDP CH CH Formyl- CH CH Aspartate CH ADP HC C HC C OHC C H4folate C C Pi N Pi N H O N N N ADP C N GTP N 2 N H N H N N H4folate 2 Fumarate 2 P H N H i ATP 2 Ribose-P Ribose-P Ribose-P Ribose-P Ribose-P Ribose-P CLE 4.3.2.2 CY 4.3.2.2 E Adenylosuccinate Inosine- 5-formamido-1-(5-P-ribosyl)- 5-amino-1-(5-P-ribosyl)- 5-amino-1-(5-P-ribosyl-) 5-amino-1-(5-P-ribosyl) ID P 2.1.2.3 T 3.5.4.10 6.3.2.6 O imidazole-4-carboxamide imidazole-4-(N)-[1,2-dicarboxy- imidazole-4-carboxylate E IMP imidazole-4-carboxamide L ethyl]-carboxamide C U N E RNA N I 2.7.7.6 2.7.7.6 R Fumarate TCA U CYCLE P RNA ACIDS RNA RNA n NUCLEIC n n+1 RNAn+1 DNA NH2 NH C 2.7.7.7 2.7.7.7 2 N O C N N C N C CH DNA DNA DNA C N CH HC C n n+1 DNAn+1 -

Inflammatory Processes
Nucleobase Deamination as a Biomarker of Inflammatory Processes by Min Dong B.S. in Pharmaceutical Science (1994) Beijing Medical University, Beijing, PR China M.S. in Biophysics (1999) Tsinghua University, Beijing, PR China SUBMITTED TO BIOLOGICAL ENGINEERING DIVISION IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THF DEGREE OF MASSACHUSETTS OF TECHNOI-OGY DOCTOR OF PHILOSOPHY FEB 2 5 at the LIBRARJIES MASSCHUSETTS INSTITUTE QF TECHNOLOGY L January 2005 © 2005 Massachusetts Institute of Technology All rights reserved Signature of Author -t oBiological EngineeringDivision January 25, 2005 Certified by Dr. Peter C. Dedon Profesr of Toxicology and Biological Engineering Am / {X7{I January 25, 2005 Acceptedby //~~~~/ ' "<~ Dr. AlanJ. Grodzinsky /m ia , Committee on Graduate Students, Biological Engineering Division AfM!CHIVES This doctoral thesis has been examined by a committee as follows: Professor Steven R. Tannenbaum Chairman / P essor Gerald N. Wogan / , Professor 'Killiam G. Thilly ...... ... ProfessofBruce Demple Dr. John S. Wishnok Professor Peter C. Dedon ............................................. Thesis Advisor -2- Nucleobase Deamination as a Biomarker of Inflammatory Processes by Min Dong SUBMITTED TO THE BIOLOGICAL ENGINEERING DIVISION IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN MOLECULAR TOXICOLOGY ABSTRACT The objective of this thesis project was to develop nucleobase deamination products as biomarkers of inflammation and to study the role of these DNA lesions in the pathophysiology -

(12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur Et Al
US 2012026.6329A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur et al. (43) Pub. Date: Oct. 18, 2012 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/10 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/24 (2006.01) CI2N 9/02 (2006.01) (75) Inventors: Eric J. Mathur, Carlsbad, CA CI2N 9/06 (2006.01) (US); Cathy Chang, San Marcos, CI2P 2L/02 (2006.01) CA (US) CI2O I/04 (2006.01) CI2N 9/96 (2006.01) (73) Assignee: BP Corporation North America CI2N 5/82 (2006.01) Inc., Houston, TX (US) CI2N 15/53 (2006.01) CI2N IS/54 (2006.01) CI2N 15/57 2006.O1 (22) Filed: Feb. 20, 2012 CI2N IS/60 308: Related U.S. Application Data EN f :08: (62) Division of application No. 1 1/817,403, filed on May AOIH 5/00 (2006.01) 7, 2008, now Pat. No. 8,119,385, filed as application AOIH 5/10 (2006.01) No. PCT/US2006/007642 on Mar. 3, 2006. C07K I4/00 (2006.01) CI2N IS/II (2006.01) (60) Provisional application No. 60/658,984, filed on Mar. AOIH I/06 (2006.01) 4, 2005. CI2N 15/63 (2006.01) Publication Classification (52) U.S. Cl. ................... 800/293; 435/320.1; 435/252.3: 435/325; 435/254.11: 435/254.2:435/348; (51) Int. Cl. 435/419; 435/195; 435/196; 435/198: 435/233; CI2N 15/52 (2006.01) 435/201:435/232; 435/208; 435/227; 435/193; CI2N 15/85 (2006.01) 435/200; 435/189: 435/191: 435/69.1; 435/34; CI2N 5/86 (2006.01) 435/188:536/23.2; 435/468; 800/298; 800/320; CI2N 15/867 (2006.01) 800/317.2: 800/317.4: 800/320.3: 800/306; CI2N 5/864 (2006.01) 800/312 800/320.2: 800/317.3; 800/322; CI2N 5/8 (2006.01) 800/320.1; 530/350, 536/23.1: 800/278; 800/294 CI2N I/2 (2006.01) CI2N 5/10 (2006.01) (57) ABSTRACT CI2N L/15 (2006.01) CI2N I/19 (2006.01) The invention provides polypeptides, including enzymes, CI2N 9/14 (2006.01) structural proteins and binding proteins, polynucleotides CI2N 9/16 (2006.01) encoding these polypeptides, and methods of making and CI2N 9/20 (2006.01) using these polynucleotides and polypeptides. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

NUCLEOSIDE CATABOLIZING ACTIVITIES in SELECTED SEEDS by Nogood Almarwani a Thesis Submitted to the Faculty of the College Of
NUCLEOSIDE CATABOLIZING ACTIVITIES IN SELECTED SEEDS By Nogood Almarwani A Thesis Submitted to the Faculty of the College of Graduate Studies at Middle Tennessee State University in Partial Fulfillment of the Requirements for the Degree of Master of Science in Chemistry Middle Tennessee State University December 2016 Thesis Committee: Dr. Paul C. Kline, Chair Dr. Andrew Burden Dr. Anthony Farone I dedicate this research work to my husband, parents, my sisters, and my brothers. ii ACKNOWLEDGMENTS This research work would not be possible without the support of a great people around me. In the beginning, I would like to thank my loving husband, Fahad Almarwani, for his endless encouragement and support me. I would also like to thank Dr. Paul C. Kline, my major professor. I cannot convey how much I appreciate him for his help and support me. Also, I would like to thank Dr. Anthony Farone and Dr. Andrew Burden as my committee members. I am grateful for their invaluable suggestions and for taking time to read my thesis. In addition, I would like to thank all the staff and faculty of the Department of Chemistry for their help and support throughout my stay at MTSU. Finally, special thanks to my parents, my sisters, my brothers, my little son, Sultan, and my friends. I would not have an opportunity to achieve this dream without their support and love. iii ABSTRACT The efficacy of pyrimidine and purine nucleotide metabolism plays a critical role in the progression of biological systems. The process of nucleotide degradation exists in all organisms. The nucleoside degradation and salvage of nucleotides, nucleosides, and nucleobases require several enzymes including deaminases and nucleosidases. -

United States Patent (19) 11 Patent Number: 4,945,049 Hamaya Et Al
United States Patent (19) 11 Patent Number: 4,945,049 Hamaya et al. (45) Date of Patent: Jul. 31, 1990 (54) METHOD FOR PREPARING MAGNETIC 0187.192 11/1983 Japan ................................... 435/168 POWDER 60-172288 9/1985 Japan. 2055092 3/1987 Japan ................................... 435/168 75) Inventors: Toru Hamaya, 1-D, Daiichiseifuso, 62-171688 7/1987 Japan. 5-5, Minami 1-chome, Meguro-ku, 62-294089 12/1987 Japan. Tokyo 152; Koki Horikoshi, 39-8, 2192870 4/1988 United Kingdom ................ 435/168 Sakuradai 4-chome, Nerima-ku, Primary Examiner-Herbert J. Lilling Tokyo 176, both of Japan Attorney, Agent, or Firm-Oblon, Spivak, McClelland, 73 Assignees: Research Development Corporation; Maier & Neustadt Toru Hamaya; Koki Horikoshi, all of Tokyo, Japan; a part interest 57 ABSTRACT The present invention relates to a method for preparing 21 Appl. No.: 343,263 magnetic powder comprising homogeneous and fine 22 PCT Filed: Aug. 18, 1988 particles using an alkali-producing enzyme. The object of the present invention is to provide a method suitable (86 PCT No.: PCT/JP88/00814 for preparing magnetic powder comprising relatively S371 Date: Apr. 14, 1989 small particles, for instance, fine particles having a par ticle size ranging from 50 to 500 nm. The present inven S 102(e) Date: Apr. 14, 1989 tion relates to a method for preparing at least one mem (87. PCT Pub. No.: WO89/01521 ber selected from the group consisting of iron oxides, iron hydroxides and iron oxyhydroxides which com PCT Pub. Date: Feb. 23, 1989 prises the step of alkalizing a solution containing iron 30 Foreign Application Priority Data ions utilizing an alkali-producing enzyme and a sub Aug. -

(12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur Et Al
US 20150240226A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur et al. (43) Pub. Date: Aug. 27, 2015 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/16 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/02 (2006.01) CI2N 9/78 (2006.01) (71) Applicant: BP Corporation North America Inc., CI2N 9/12 (2006.01) Naperville, IL (US) CI2N 9/24 (2006.01) CI2O 1/02 (2006.01) (72) Inventors: Eric J. Mathur, San Diego, CA (US); CI2N 9/42 (2006.01) Cathy Chang, San Marcos, CA (US) (52) U.S. Cl. CPC. CI2N 9/88 (2013.01); C12O 1/02 (2013.01); (21) Appl. No.: 14/630,006 CI2O I/04 (2013.01): CI2N 9/80 (2013.01); CI2N 9/241.1 (2013.01); C12N 9/0065 (22) Filed: Feb. 24, 2015 (2013.01); C12N 9/2437 (2013.01); C12N 9/14 Related U.S. Application Data (2013.01); C12N 9/16 (2013.01); C12N 9/0061 (2013.01); C12N 9/78 (2013.01); C12N 9/0071 (62) Division of application No. 13/400,365, filed on Feb. (2013.01); C12N 9/1241 (2013.01): CI2N 20, 2012, now Pat. No. 8,962,800, which is a division 9/2482 (2013.01); C07K 2/00 (2013.01); C12Y of application No. 1 1/817,403, filed on May 7, 2008, 305/01004 (2013.01); C12Y 1 1 1/01016 now Pat. No. 8,119,385, filed as application No. PCT/ (2013.01); C12Y302/01004 (2013.01); C12Y US2006/007642 on Mar. 3, 2006.