Reconstruction of Metabolic Pathways for the Cattle Genome Seongwon Seo1 and Harris a Lewin*1,2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mycobacterium Haemophilum Sp. Nov., a New Pathogen of Humanst
0020-7713/78/0028-0067$02.0/0 INTERNATIONALJOURNAL OF SYSTEMATICBACTERIOLOGY, Jan. 1978, p. 67-75 Vol. 28, No. 1 Copyright 0 1978 International Association of Microbiological Societies Printed in U.S. A. Mycobacterium haemophilum sp. nov., a New Pathogen of Humanst DAVID SOMPOLINSKY,’v2 ANNIE LAGZIEL,’ DAVID NAVEH,3 AND TULI YANKILEVITZ3 Department of Microbwlogy, Asaf Harofe Government Hospital, Zerifin‘; Rapaport Laboratories, Bar-Ilan University, Ramat-Gan2;and Department of Internal Medicine “A,”Meir Hospital, Kfar Saba,3 Israel A patient under immunosuppressive treatment of Hodgkin’s disease developed generalized skin granulomata and subcutaneous abscesses. Several aspirated pus samples yielded acid-fast rods with the following properties: temperature opti- mum, about 30°C with no growth at 37°C; slow growth (2 to 4 weeks); nonchrom- ogenic; hemoglobin or hemin requirement for growth; catalase negative; pyrazin- amidase and nicotinamidase positive; and urease negative. The guanine-plus- cytosine content of the deoxyribonucleic acid was calculated from the melting temperature to be 66.0 mol%. It is concluded that these isolates belong to a new species, for which the name Mycobacterium haemophilum is proposed. The type strain of this species is strain 1 (= ATCC 29548). The new species is related to M. marinum and M. ulcerans. Granulomatous skin diseases of humans CASE HISTORY caused by mycobacteria other than Mycobacte- After World War 11, a 27-year-old woman was di- rium tuberculosis and M. leprae are well known. agnosed as having tuberculosis. She received treat- The two organisms most often involved etiolog- ment until 1951. In February 1969, at the age of 51, ically are M. -

Calorie Restriction and the Exercise of Chromatin
Downloaded from genesdev.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Calorie restriction and the exercise of chromatin Alejandro Vaquero1,4 and Danny Reinberg2,3 1Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), ICREA, and IDIBELL, L’Hospitalet de Llobregat, Barcelona 08907, Spain; 2Howard Hughes Medical Institute, Department of Biochemistry, New York University-Medical School, New York, New York 10016, USA Since the earliest stages of evolution, organisms have (Masoro2005).Inahallmarkstudyin1935,McCayetal. faced the challenge of sensing and adapting to environ- (1935) demonstrated that, compared with rats fed with mental changes for their survival under compromising a standard diet, rats fed under CR lived longer, weighed less, conditions such as food depletion or stress. Implicit in showed heart hypertrophy, and had smaller livers, which these responses are mechanisms developed during evolu- they explained at the time as an effect of growth retarda- tion that include the targeting of chromatin to allow or tion. Since then, considerable research efforts have revealed prevent expression of fundamental genes and to protect that, surprisingly, the effects of CR on life span are highly genome integrity. Among the different approaches to similar among diverse eukaryotes (e.g., yeast, insects, fish, study these mechanisms, the analysis of the response to and mammals) (Kennedy et al. 2007). Although we can only a moderate reduction of energy intake, also known as speculate on these findings, they suggest that the follow- calorie restriction (CR), has become one of the best sources ing general survival strategy has been conserved through- of information regarding the factors and pathways in- out eukaryotic evolution: In low-nutrient conditions, volved in metabolic adaptation from lower to higher metabolism is adjusted to enable more efficient use of eukaryotes. -

Metallochaperones Are Needed for Mycobacterium Tuberculosis And
RESEARCH ARTICLE crossm Metallochaperones Are Needed for Mycobacterium tuberculosis and Escherichia coli Nicotinamidase-Pyrazinamidase Activity Downloaded from Patricia Sheen,a Anuntxi Monsalve,a Jhanina Campos,a Rodolfo Huerta,a Ricardo Antiparra,a Héctor Arteaga,a Patricia Duran,a Carlos Bueno,a Daniela E. Kirwan,b Robert H. Gilman,c Mirko Zimica aLaboratory of Bioinformatics and Molecular Biology, Department of Cellular and Molecular Sciences, School of Science, Universidad Peruana Cayetano Heredia, Lima, Peru bInfection and Immunity Research Institute, St. George’s, University of London, London, United Kingdom c Department of International Health, School of Public Health, Johns Hopkins University, Baltimore, Maryland, USA http://jb.asm.org/ ABSTRACT Mycobacterium tuberculosis nicotinamidase-pyrazinamidase (PZAse) is a metalloenzyme that catalyzes conversion of nicotinamide-pyrazinamide to nicotinic acid-pyrazinoic acid. This study investigated whether a metallochaperone is required for optimal PZAse activity. M. tuberculosis and Escherichia coli PZAses (PZAse-MT and PZAse-EC, respectively) were inactivated by metal depletion (giving PZAse-MT–Apo and PZAse-EC–Apo). Reactivation with the E. coli metallochaperone ZnuA or Rv2059 (the M. tuberculosis analog) was measured. This was repeated following proteolytic on January 16, 2020 at ST GEORGE'S LIBRARY and thermal treatment of ZnuA and Rv2059. The CDC1551 M. tuberculosis reference strain had the Rv2059 coding gene knocked out, and PZA susceptibility and the pyr- azinoic acid (POA) efflux rate were measured. ZnuA (200 M) achieved 65% PZAse- EC–Apo reactivation. Rv2059 (1 M) and ZnuA (1 M) achieved 69% and 34.3% PZAse-MT–Apo reactivation, respectively. Proteolytic treatment of ZnuA and Rv2059 and application of three (but not one) thermal shocks to ZnuA significantly reduced the capacity to reactivate PZAse-MT–Apo. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Appl. Microbiol. Biotechnol. 78: 293-307
Appl Microbiol Biotechnol (2008) 78:293–307 DOI 10.1007/s00253-007-1308-y APPLIED GENETICS AND MOLECULAR BIOTECHNOLOGY PA2663 (PpyR) increases biofilm formation in Pseudomonas aeruginosa PAO1 through the psl operon and stimulates virulence and quorum-sensing phenotypes Can Attila & Akihiro Ueda & Thomas K. Wood Received: 19 October 2007 /Revised: 26 November 2007 /Accepted: 28 November 2007 / Published online: 22 December 2007 # Springer-Verlag 2007 Abstract Previously, we identified the uncharacterized PA2130, and PA4549) and for small molecule transport predicted membrane protein PA2663 of Pseudomonas (PA0326, PA1541, PA1632, PA1971, PA2214, PA2215, aeruginosa PAO1 as a virulence factor using a poplar tree PA2678, and PA3407). Phenotype arrays also showed that model; PA2663 was induced in the poplar rhizosphere and, PA2663 represses growth on D-gluconic acid, D-mannitol, upon inactivation, it caused 20-fold lower biofilm forma- and N-phthaloyl-L-glutamic acid. Hence, the PA2663 gene tion (Attila et al., Microb Biotechnol, 2008). Here, we product increases biofilm formation by increasing the psl- confirmed that PA2663 is related to biofilm formation by operon-derived exopolysaccharides and increases pyover- restoring the wild-type phenotype by complementing the dine synthesis, PQS production, and elastase activity while PA2663 mutation in trans and investigated the genetic basis reducing swarming and swimming motility. We speculate of its influence on biofilm formation through whole- that PA2663 performs these myriad functions as a novel transcriptome and -phenotype studies. Upon inactivating membrane sensor. PA2663 by transposon insertion, the psl operon that encodes a galactose- and mannose-rich exopolysaccharide Keywords Biofilm . Pseudomonas aeruginosa . was highly repressed (verified by RT-PCR). -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_004758865Cyc: Acinetobacter baumannii CIAT758 Cellular Overview Connections between pathways are omitted for legibility. -

Table 4. V. Cholerae Flexgene ORF Collection
Table 4. V. cholerae FLEXGene ORF collection Reference Clone protein PlasmID clone GenBank Locus tag Symbol accession identifier FLEX clone name accession Product name VC0001 NP_062585 VcCD00019918 FLH200476.01F DQ772770 hypothetical protein VC0002 mioC NP_062586 VcCD00019938 FLH200506.01F DQ772771 mioC protein VC0003 thdF NP_062587 VcCD00019958 FLH200531.01F DQ772772 thiophene and furan oxidation protein ThdF VC0004 yidC NP_062588 VcCD00019970 FLH200545.01F DQ772773 inner membrane protein, 60 kDa VC0005 NP_062589 VcCD00061243 FLH236482.01F DQ899316 conserved hypothetical protein VC0006 rnpA NP_062590 VcCD00025697 FLH214799.01F DQ772774 ribonuclease P protein component VC0007 rpmH NP_062591 VcCD00061229 FLH236450.01F DQ899317 ribosomal protein L34 VC0008 NP_062592 VcCD00019917 FLH200475.01F DQ772775 amino acid ABC transporter, ATP-binding protein VC0009 NP_062593 VcCD00019966 FLH200540.01F DQ772776 amino acid ABC transproter, permease protein VC0010 NP_062594 VcCD00019152 FLH199275.01F DQ772777 amino acid ABC transporter, periplasmic amino acid-binding portion VC0011 NP_062595 VcCD00019151 FLH199274.01F DQ772778 hypothetical protein VC0012 dnaA NP_062596 VcCD00017363 FLH174286.01F DQ772779 chromosomal DNA replication initiator DnaA VC0013 dnaN NP_062597 VcCD00017316 FLH174063.01F DQ772780 DNA polymerase III, beta chain VC0014 recF NP_062598 VcCD00019182 FLH199319.01F DQ772781 recF protein VC0015 gyrB NP_062599 VcCD00025458 FLH174642.01F DQ772782 DNA gyrase, subunit B VC0016 NP_229675 VcCD00019198 FLH199346.01F DQ772783 hypothetical protein -

Purine and Pyrimidine Metabolism in Human Epidermis* Jean De Bersaques, Md
THE JOURNAL OP INVESTIGATIVE DERMATOLOGY Vol. 4s, No. Z Copyright 1957 by The Williams & Wilkins Co. Fri nte,1 in U.S.A. PURINE AND PYRIMIDINE METABOLISM IN HUMAN EPIDERMIS* JEAN DE BERSAQUES, MD. The continuous cellular renewal occurring inthine, which contained 5% impurity, and for uric the epidermis requires a very active synthesisacid, which consisted of 3 main components. The reaction was stopped after 1—2 hours in- and breakdown of nuclear and cytoplasmiecubation at 37° and the products were spotted on nucleic acids. Data on the enzyme systemsWhatman 1 filter paper sheets. According to the participating in these metabolic processes arereaction products expected, a choice was made of rather fragmentary (1—9) and some are, inat least 2 among the following solvents, all used terms of biochemical time, in need of up- in ascending direction: 1. isoamyl alcohol—5% Na2HPO4 (1:1), dating. In some other publications (10—18), 2. water-saturated n-butanol, the presence and concentration of various in- 3. distilled water, termediate products is given. 4. 80% formic acid—n-hutanol——n-propanol— In this paper, we tried to collect and supple- acetone—30% trichloro-aeetic acid (5:8:4: ment these data by investigating the presence 5:3), 5. n-butanol——4% boric acid (43:7), or absence in epidermis of enzyme systems 6. isobutyrie acid—water—ammonia 0.880—ver- that have been described in other tissues. sene 0.1M(500:279:21:8), This first investigation was a qualitative one, 7. upper phase of ethyl acetate—water—formic and some limitations were set by practical acid (12:7:1), 8. -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -

Deep Sequencing of the Camellia Sinensis Transcriptome Revealed
Shi et al. BMC Genomics 2011, 12:131 http://www.biomedcentral.com/1471-2164/12/131 RESEARCHARTICLE Open Access Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds Cheng-Ying Shi1†, Hua Yang1†, Chao-Ling Wei1†, Oliver Yu1,2, Zheng-Zhu Zhang1, Chang-Jun Jiang1, Jun Sun1, Ye-Yun Li1, Qi Chen1, Tao Xia1, Xiao-Chun Wan1* Abstract Background: Tea is one of the most popular non-alcoholic beverages worldwide. However, the tea plant, Camellia sinensis, is difficult to culture in vitro, to transform, and has a large genome, rendering little genomic information available. Recent advances in large-scale RNA sequencing (RNA-seq) provide a fast, cost-effective, and reliable approach to generate large expression datasets for functional genomic analysis, which is especially suitable for non-model species with un-sequenced genomes. Results: Using high-throughput Illumina RNA-seq, the transcriptome from poly (A)+ RNA of C. sinensis was analyzed at an unprecedented depth (2.59 gigabase pairs). Approximate 34.5 million reads were obtained, trimmed, and assembled into 127,094 unigenes, with an average length of 355 bp and an N50 of 506 bp, which consisted of 788 contig clusters and 126,306 singletons. This number of unigenes was 10-fold higher than existing C. sinensis sequences deposited in GenBank (as of August 2010). Sequence similarity analyses against six public databases (Uniprot, NR and COGs at NCBI, Pfam, InterPro and KEGG) found 55,088 unigenes that could be annotated with gene descriptions, conserved protein domains, or gene ontology terms. Some of the unigenes were assigned to putative metabolic pathways. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -
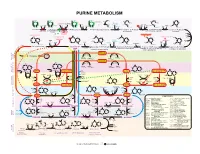
15 Purines Apr13.Ai
PURINE METABOLISM CH O-P CH O-P CH O-P CH O-P NH NH NH NH 2 2 2 2 H C 2 H C H C HC O H O H O NH O NHCOCH NH 2 2 2 2 2 2 CHO CHO CH Formyl- Glutamine Glutamate OC OC C C H H H H H H Glycine H H or NH H folate NH ADP N H OH H H H H 4 ATP Pi HN = NH ADP ATP A MP O-P-O-P H O H ATP ADP+P Gln H O Glu H N 2 PP H folate 2 ATP Pi 2 OH OH OH OH OH OH OH OH Ribose-P 4 Ribose-P Ribose-P Ribose-P 5-P-ribose 5-P-ribosyl- 5-P-Ribosylamine N1-(5-P-ribosyl)glycinamide N2-formyl-N1-(5-P-ribosyl) 2-(formamido)-N1- 5-amino-1-(P-ribosyl)- 2.7.6.1 pyrophosphate 2.4.2.14 6.3.4.13 2.1.2.2 6.3.5.3 (5-P-ribosyl)acetamidine 6.3.3.1 ADP GDP AMP GTP glycinamide - imidazole (PRPP) GDP ADP ATP ATPGMP -OOCCHCH COO- -OOCCHCH COO- 2 2 O O O CO NH - - - - 2 OOCCHCH COO C HN CO OOCCHCH2COO - C NH 2 C N C NH NH NH OOC 4.1.1.21 N 2 HN H N + NH N C C H2N C 2 C C Aspartate NH3 C CH GDP CH CH Formyl- CH CH Aspartate CH ADP HC C HC C OHC C H4folate C C Pi N Pi N H O N N N ADP C N GTP N 2 N H4folate H N H N N 2 Fumarate 2 Pi ATP H N Ribose-P Ribose-P H Ribose-P Ribose-P Ribose-P 2 E 4.3.2.2 Ribose-P YCL C Adenylosuccinate 4.3.2.2 E Inosine- 5-formamido-1-(5-P-ribosyl)- 5-amino-1-(5-P-ribosyl)- 5-amino-1-(5-P-ribosyl-) 5-amino-1-(5-P-ribosyl) ID P 3.5.4.10 2.1.2.3 T i m i d a z ole-4-(N)-[1,2-dicarboxy-6.3.2.6 O imidazole-4-carboxamide imidazole-4-carboxylate E IMP imidazole-4-carboxamide L ethyl]-carboxamide C U N C E I RNA N S E I 2.7.7.6 2.7.7.6 R D L TCA I Fumarate U C C CYCLE P U RNA A RNA RNA n N n n+1 RNAn+1 DNA S NH2 E 2.7.7.7 2.7.7.7 E NH C T 2 N O D A C I N N C H T N