Quadrisol Oral Gel for Dogs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Horsemen's Information 2016和文TGP 1 薬物修正後 E
[Conditions] 1 Date December 29 (Tue), 2020 2020 Oi Racetrack, Race 10 2 Location TCK, Oi Racetrack 3 Race The 66th Running of Tokyo Daishoten (GI) 4 Eligibility Thoroughbreds, 3 years old & up 5 Full Gate 16 horses 6 Foreign Runners Selected by the selection committee from among the pre-entered horses. 7 Distance 2,000m, 1 1/4 mile (Right-handed, dirt course) 8 Weight 3 years old: 121.5 lbs,4 years old & up: 125.5 lbs, Female: 4.4 lbs less For 3 year-old-horses from the southern hemisphere, reduce 4.4 lbs from the above weight. 9 Purse Unit: 1,000 JPY Prize Purse & Bonus money Running Record 1st place 6th place allowances prize *1 prize *2 1st place 2nd place 3rd place 4th place 5th place or lower Owner 80,000 28,000 16,000 8,000 4,000 300 300 50 1,600 Trainer 880 70 60 50 40 30 30 80 Jockey 120 110 100 80 70 60 30 80 Groom 80 70 60 50 40 30 30 80 Rider 30 30 80 *1 Paid for the runner who broke the previous record and also set the best record during the race. *2 Prize equivalent to the amount listed in the table above is presented. *3 1 USD= 105.88 JPY (As of August,2020) 10 Handling of Late Scratch No allowance is paid in the case of a late scratch (including cancelation of race due to standstill in a starting gate) approved by TCK, Stewards, and Starter. However, if the chairman of the race meeting operation committee deems that the horse is involved in an accident not caused by the horse, the owner is given an running allowance. -

Survey of Pain Knowledge and Analgesia in Dogs and Cats by Colombian Veterinarians
veterinary sciences Article Survey of Pain Knowledge and Analgesia in Dogs and Cats by Colombian Veterinarians Carlos Morales-Vallecilla 1, Nicolas Ramírez 1, David Villar 1,*, Maria Camila Díaz 1 , Sandra Bustamante 1 and Duncan Ferguson 2 1 Facultad de Ciencias Agrarias Universidad de Antioquia, Medellín 050010, Colombia; [email protected] (C.M.-V.); [email protected] (N.R.); [email protected] (M.C.D.); [email protected] (S.B.) 2 Department of Comparative Biosciences, College of Veterinary Medicine, University of Illinois at Urbana-Champaign, Urbana, IL 61802, USA; [email protected] * Correspondence: [email protected]; Tel.: +57-3178047381 Received: 6 December 2018; Accepted: 5 January 2019; Published: 10 January 2019 Abstract: A questionnaire study was conducted among 131 veterinarians practicing in the city of Medellin, Colombia, to assess views on pain evaluation and management in dogs and cats. When pain recognition and quantification abilities were used as a perceived competence of proper pain assessment, only 83/131 (63.4%, confidence interval (CI) 0.55–0.72) were deemed to have satisfactory skills, with the rest considered to be deficient. There were 49/131 (37.4) veterinarians who had participated in continuing education programs and were more confident assessing pain, with an odds ratio ( standard error) of 2.84 1.15 (p = 0.01; CI 1.27–6.32). In addition, the odds of using ± ± pain scales was 4.28 2.17 (p < 0.01, CI 1.58–11.55) greater if they had also participated in continuing ± education programs. The term multimodal analgesia was familiar to 77 (58.7%) veterinarians who also claimed to use more than one approach to pain control. -
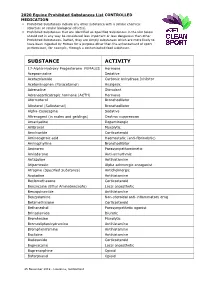
2020 Equine Prohibited Substances List CONTROLLED MEDICATION
2020 Equine Prohibited Substances List CONTROLLED MEDICATION . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). Prohibited Substances that are identified as Specified Substances in the List below should not in any way be considered less important or less dangerous than other Prohibited Substances. Rather, they are simply substances which are more likely to have been ingested by Horses for a purpose other than the enhancement of sport performance, for example, through a contaminated food substance. SUBSTANCE ACTIVITY 17-Alpha-Hydroxy Progesterone FEMALES Hormone Acepromazine Sedative Acetazolamide Carbonic Anhydrase Inhibitor Acetominophen (Paracetamol) Analgesic Adrenaline Stimulant Adrenocorticotropic hormone (ACTH) Hormone Aformoterol Bronchodilator Albuterol (Salbutamol) Bronchodilator Alpha-Casozepine Sedative Altrenogest (in males and geldings) Oestrus suppression Amantadine Dopaminergic Ambroxol Mucolytic Amcinonide Corticosteroid Aminocaproic acid Haemostatic (anti-fibrinolytic) Aminophylline Bronchodilator Aminorex Parasympathomimetic Amiodarone Anti-arrhythmic Antazoline Antihistamine Atipamezole Alpha adrenergic antagonist Atropine (Specified Substance) Anticholinergic Azatadine Antihistamine Beclomethasone Corticosteroid Benzocaine (Ethyl Aminobenzoate) Local anaesthetic Benzquinamide Antihistamine Benzydamine Non-steroidal anti-inflammatory drug Betamethasone Corticosteroid Bethanechol Parasympathetic agonist Brinzolamide Diuretic Bromhexine Mucolytic Bromodiphenhydramine -

An End-To-End Workflow for Quantitative Screening of Multiclass, Multiresidue Veterinary Drugs in Meat Using the Agilent 6470 Triple Quadrupole LC/MS
Application Note Food Testing & Agriculture An End-To-End Workflow for Quantitative Screening of Multiclass, Multiresidue Veterinary Drugs in Meat Using the Agilent 6470 Triple Quadrupole LC/MS Authors Abstract Siji Joseph, Aimei Zou, Chee A comprehensive LC/MS/MS workflow was developed for targeted screening or Sian Gan, Limian Zhao, and quantitation of 210 veterinary drug residues in animal muscle prepared for human Patrick Batoon consumption, with the intention to accelerate and simplify routine laboratory testing. Agilent Technologies, Inc. The workflow ranged from sample preparation through chromatographic separation, MS detection, data processing and analysis, and report generation. The workflow performance was evaluated using three muscle matrices—chicken, pork, and beef— and was assessed on two different Agilent triple quadrupole LC/MS models (an Agilent 6470 and a 6495C triple quadrupole LC/MS). A simple sample preparation protocol using Agilent Captiva EMR—Lipid cartridges provided efficient extraction and matrix cleanup. A single chromatographic method using Agilent InfinityLab Poroshell 120 EC-C18 columns with a 13-minute method delivered acceptable separation and retention time distribution across the elution window for reliable triple quadrupole detection and data analysis. Workflow performance was evaluated based on evaluation of limit of detection (LOD), limit of quantitation (LOQ), calibration curve linearity, accuracy, precision, and recovery, using matrix-matched spike samples for a range from 0.1 to 100 μg/L. Calibration curves were plotted from LOQ to 100 μg/L, where all analytes demonstrated linearity R2 >0.99. Instrument method accuracy values were within 73 to 113%. Target analytes response and retention time %RSD values were ≤19% and ≤0.28% respectively. -

Assessment of the Efficacy of Firocoxib and Robenacoxib
Assessment of the Efficacy of Firocoxib and Robenacoxib in an Induced Synovitis Model of Acute Arthritis in Dogs Christelle Dauteloupa Corinne Pichoub Frederic Beugneta,* aMerial SAS, 2 Av Pasteur, 69007, Lyon, France b Amatsigroup, Site AmatsiAvogadro, Parc de Génibrat, 31470 Fontenilles, France * Corresponding author: E-mail address: [email protected] KEY WORDS: Firocoxib, Robenacoxib, and the increased PVF values compared to Arthritis, Lameness score, Peak Vertical the control group at 3 and 5 hours post- force injection. Firocoxib performed significantly better than robenacoxib at 3, 5, and 10 hours ABSTRACT post-UC injection. In this model, robena- The objective of this study was to compare coxib was not different from control for both the analgesic activity of a single oral dose the VLS and the PVF values. Pre-treatment of two COX-2 selective inhibitors, firo- with firocoxib reduced the induced pain as- coxib (Previcox®, Merial) and robenacoxib sociated with intra-articular administration (Onsior®, Elanco), in an acute pain model of urate crystals. in dogs. Sixteen healthy Beagle dogs were randomly allocated to three groups. Two INTRODUCTION successive experiments were conducted, in Chronic osteoarthritis is common in dogs which eight dogs served as control. Eight and is estimated to affect 20% of dogs dogs received firocoxib or robenacoxib in a over 1 year of age (Johnston, 1997). Since cross-over design at the recommended dos- no drug has been shown to reverse the age 13 hours before intra-articular injection pathological changes of osteoarthritis, the of a urate crystal suspension (UC) for induc- objective of treatment is to reduce pain and tion of synovitis. -

A. List of Prohibited Substances 1
Reviewed 09.05.17 List over prohibited substances and withdrawal times in Scandinavia, valid from May 20th, 2017 This list has been developed in collaboration with the other Scandinavian countries through NEMAC (Nordic Equine Medication and Anti-doping Committee) The List of prohibited substances and withdrawal times consists of the A-list, listing substances and treatment methods that are absolutely prohibited for horses, and the B-list, listing substances that are prohibited in competition; the withdrawal times for these substances as well as treatment methods with withdrawal times. The list may be reviewed several times per year. This list is valid starting from May 20th, 2017 and is enforced until a new list takes effect. A valid list of withdrawal times can also be found at any time on DNT’s official website, www.travsport.no.ST’s official website, www.travsport.se, and DTC`s official website www.trav.dk Health certificate and keeping of medical records The trainer is responsible for ensuring that any treatment that requires a withdrawal time is listed in the horse’s health certificate. The start and end dates of the treatment, the name of the treatment/medication/active substance, amount given, method of administration, withdrawal time as well as the name of the veterinarian or other person responsible for the treatment must all be listed in the health certificate in accordance with DNT’s Doping Regulations 2017 § 4. Passport and health certificate should be brought with the horse at all times. Omitting, incompletely or improperly listing treatments in the health certificate constitutes a breach of the Doping Regulations. -

Chromatography of Non-Steroidal Anti-Inflammatory Drugs
A tica nal eu yt c ic a a m A r a c t Szogyi and Cserháti, Pharm Anal Acta 2012, 3:2 h a P DOI: 10.4172/2153-2435.1000149 ISSN: 2153-2435 Pharmaceutica Analytica Acta Review Article Open Access Chromatography of Non-Steroidal Anti-Inflammatory Drugs: New Achievements Mária Szőgyi* and Tibor Cserháti Institute of Material and Environmental Chemistry, Hungarian Academy of Sciences, Po Box-17, H-1525 Budapest, Hungary Abstract The newest results in the chromatographic analysis of non-steroidal anti-inflammatory drugs present in various organic and inorganic accompanying matrices are collected and critically evaluated. Examples for the application of pre-concentration and pre-purification methods for the enhancement of the selectivity and sensitivity of chromatographic technologies are presented. The advantages and disadvantages of the different chromatographic methods are discussed and the separation capacities of the techniques are compared. The application of various chromatographic procedures for the separation and quantitative determination of non-steroidal anti-inflammatory drug (ibuprofen, diclofenac, genfibrozil, ketoprofen, diflunisal, indoprofen, fenoprofen, meclofenamic acid, naproxen, mexiletin) is discussed in detail. Keywords: Non-steroidal anti-inflammatory drugs; Gas The objectives of the present review are the compilation and concise chromatography; Liquid chromatographic technologies; Electrically evaluation on the newest results obtained in the chromatographic driven separation methods analysis of NSAIDs present in human -

Recognition and Alleviation of Pain in Laboratory Animals (2009)
THE NATIONAL ACADEMIES PRESS This PDF is available at http://nap.edu/12526 SHARE Recognition and Alleviation of Pain in Laboratory Animals (2009) DETAILS 198 pages | 6 x 9 | PAPERBACK ISBN 978-0-309-12834-6 | DOI 10.17226/12526 CONTRIBUTORS GET THIS BOOK Committee on Recognition and Alleviation of Pain in Laboratory Animals; Institute for Laboratory Animal Research; Division on Earth and Life Studies; National Research Council FIND RELATED TITLES SUGGESTED CITATION National Research Council 2009. Recognition and Alleviation of Pain in Laboratory Animals. Washington, DC: The National Academies Press. https://doi.org/10.17226/12526. Visit the National Academies Press at NAP.edu and login or register to get: – Access to free PDF downloads of thousands of scientific reports – 10% off the price of print titles – Email or social media notifications of new titles related to your interests – Special offers and discounts Distribution, posting, or copying of this PDF is strictly prohibited without written permission of the National Academies Press. (Request Permission) Unless otherwise indicated, all materials in this PDF are copyrighted by the National Academy of Sciences. Copyright © National Academy of Sciences. All rights reserved. Recognition and Alleviation of Pain in Laboratory Animals Recognition and Alleviation of Pain in Laboratory Animals Committee on Recognition and Alleviation of Pain in Laboratory Animals Institute for Laboratory Animal Research Division on Earth and Life Studies Copyright National Academy of Sciences. All rights reserved. Recognition and Alleviation of Pain in Laboratory Animals THE NATIONAL ACADEMIES PRESS 500 Fifth Street, NW Washington, DC 20001 NOTICE: The project that is the subject of this report was approved by the Govern- ing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineer- ing, and the Institute of Medicine. -

And Grapiprant (Galliprant®) in an Induced Model of Acute Arthritis in Dogs Andrea García De Salazar Alcalá1, Lucile Gioda1, Alia Dehman2 and Frederic Beugnet3*
Salazar Alcalá et al. BMC Veterinary Research (2019) 15:309 https://doi.org/10.1186/s12917-019-2052-0 RESEARCH ARTICLE Open Access Assessment of the efficacy of firocoxib (Previcox®) and grapiprant (Galliprant®) in an induced model of acute arthritis in dogs Andrea García de Salazar Alcalá1, Lucile Gioda1, Alia Dehman2 and Frederic Beugnet3* Abstract Background: Non-steroidal anti-inflammatory drugs (NSAIDs) are an important tool in the management of canine osteoarthritis, with the most recent introduction into the category being grapiprant, a piprant that selectively targets the EP4 prostaglandin receptor. To date there have been no efficacy studies comparing grapiprant with other NSAIDs. A randomized, two-sequence, assessor-blinded study involving two separate experiments was undertaken to measure the potency and persistence of acute pain control over 24 h resulting from a single oral dose of either firocoxib (Previcox®) or grapiprant (Galliprant®) in an acute arthritis model. Results: Force-plate derived lameness ratios (0, no force recorded on the plate; 1, normal force) for the untreated group remained at 0 for most post-arthritis induction (PAI) assessments in both experiments. Throughout Experiment 1, mean PAI lameness ratios of the firocoxib-treated group remained at or above 0.80. In the grapiprant-treated group, ratios were 0 at 5 and 7 h PAI (7 and 9 h post-treatment), and 0.16 at 10 h PAI (12 h post-treatment). For lameness ratios, relative to the firocoxib group, the control and grapiprant group ratios were significantly lower at each PAI assessment (p ≤ 0.026 and p < 0.001, respectively), except at 1.5 h PAI at which acute pain was still not installed in untreated control dogs. -

WO 2013/020527 Al 14 February 2013 (14.02.2013) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2013/020527 Al 14 February 2013 (14.02.2013) P O P C T (51) International Patent Classification: (74) Common Representative: UNIVERSITY OF VETER¬ A61K 9/06 (2006.01) A61K 47/32 (2006.01) INARY AND PHARMACEUTICAL SCIENCES A61K 9/14 (2006.01) A61K 47/38 (2006.01) BRNO FACULTY OF PHARMACY; University of A61K 47/10 (2006.01) A61K 9/00 (2006.01) Veterinary and Pharmaceutical Sciences Brno Faculty Of A61K 47/18 (2006.01) Pharmacy, Palackeho 1/3, CZ-61242 Brno (CZ). (21) International Application Number: (81) Designated States (unless otherwise indicated, for every PCT/CZ20 12/000073 kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (22) Date: International Filing BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 2 August 2012 (02.08.2012) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (25) Filing Language: English HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (26) Publication Language: English ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (30) Priority Data: NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, 201 1-495 11 August 201 1 ( 11.08.201 1) SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, 2012- 72 1 February 2012 (01.02.2012) TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, 2012-5 11 26 July 2012 (26.07.2012) ZW. -

Non Steroidal Anti Inflammatory Drugs (NSAIDS)
Fact Sheet Fact Sheet ChokNon-Steroidale Anti- Choke is a relatively common condition seen in horses and ponies and is typically caused by obstruction of the oesophagus (food pipe)Inflammatory with food; occasionally a foreign Drugs body can be involved e.g. wood or plastic. FortunatelyNon-steroidal anti-inflammatory drugs (NSAIDs) are probably many cases of choke resolve quickly and spontaneouslysome of the most widely used drugs used in both human and only cases in which the obstruction lasts for longerand equine medicine. They act to reduce pain by inhibiting than 30 minutes are likely to require veterinary assistancthe einflammatory. pathways after injury. Common NSAIDs It is important to note that this is not the same as the used in human medicine include aspirin, paracetamol life-threatening condition in humans, where the term and ibuprofen. NSAIDs used in equine medicine include “choke” refers to blockage of the windpipe rather thanphenylbutazone the (bute), meloxicam, suxibuzone and flunixin. oesophagus. This difference means that unlike humans, horses with choke can still breathe. NSAIDs may be administered by injection, orally (as a powder, granules or paste given in feed or by mouth) or in a cream, ointment, gel or lotion to apply to the surface of inflamed tissues such as the skin. Clinical signs: • difficulty/repeated attempts at swallowing • stretching/arching of the neck • coughing NSAIDs are a benefit to the welfare of the equine • food & saliva discharging from the nose population through the control of pain and inflammation. • drooling • disinterest in food REGULAThe term R‘non-steroidal’ DENTAL EXAMIN is usedATIONS to distinguish AND these • occasionally a lump may be seen or felt TREdrugsATMENT from C steroidAN REDU anti-inflammatoriesCE THE RISK OF (cortisone).CHOKE on the left side of the neck. -

FIROCOXIB 57 Mg VETERINARY TABLETS
FIROCOXIB 57 mg VETERINARY TABLETS Degree Final Project Daniel Cases Poley Main Scope: Pharmaceutical Technology Secondary Scopes: Biopharmacy and History and Legislation Facultat de Farmàcia i Ciències de l'Alimentació Universitat de Barcelona June 2019 Degree Final Project This work is licenced under a Creative Commons license. Degree Final Project Abstract ........................................................................................................................... 1 Scopes involved ................................................................................................................ 2 1 Introduction ................................................................................................................... 2 2 Objectives ..................................................................................................................... 2 3 Planning design ............................................................................................................. 3 4 Pharmacological view ..................................................................................................... 3 4.1 Pain ........................................................................................................................ 3 4.2 Mechanism of action and selectivity .......................................................................... 5 4.3 The clinical experience of firocoxib ........................................................................... 7 4.4 The last advances with firocoxib ............................................................................