DNA Replication Is the Target for the Antibacterial Effects of Nonsteroidal Anti-Inflammatory Drugs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent (19) 11 Patent Number: 5,955,504 Wechter Et Al
USOO5955504A United States Patent (19) 11 Patent Number: 5,955,504 Wechter et al. (45) Date of Patent: Sep. 21, 1999 54 COLORECTAL CHEMOPROTECTIVE Marnett, “Aspirin and the Potential Role of Prostaglandins COMPOSITION AND METHOD OF in Colon Cancer, Cancer Research, 1992; 52:5575–89. PREVENTING COLORECTAL CANCER Welberg et al., “Proliferation Rate of Colonic Mucosa in Normal Subjects and Patients with Colonic Neoplasms: A 75 Inventors: William J. Wechter; John D. Refined Immunohistochemical Method.” J. Clin Pathol, McCracken, both of Redlands, Calif. 1990; 43:453-456. Thun et al., “Aspirin Use and Reduced Risk of Fatal Colon 73 Assignee: Loma Linda University Medical Cancer." N Engl J Med 1991; 325:1593-6. Center, Loma Linda, Calif. Peleg, et al., “Aspirin and Nonsteroidal Anti-inflammatory Drug Use and the Risk of Subsequent Colorectal Cancer.” 21 Appl. No.: 08/402,797 Arch Intern Med. 1994, 154:394–399. 22 Filed: Mar 13, 1995 Gridley, et al., “Incidence of Cancer among Patients With Rheumatoid Arthritis J. Natl Cancer Inst 1993 85:307-311. 51) Int. Cl. .......................... A61K 31/19; A61K 31/40; Labayle, et al., “Sulindac Causes Regression Of Rectal A61K 31/42 Polyps. In Familial Adenomatous Polyposis” Gastroenterol 52 U.S. Cl. .......................... 514/568; 514/569; 514/428; ogy 1991 101:635-639. 514/416; 514/375 Rigau, et al., “Effects Of Long-Term Sulindac Therapy On 58 Field of Search ..................................... 514/568, 570, Colonic Polyposis” Annals of Internal Medicine 1991 514/569, 428, 416, 375 11.5:952-954. Giardiello.et al., “Treatment Of Colonic and Rectal 56) References Cited Adenomas With Sulindac In Familial Adenomatous Poly U.S. -

Optum Essential Health Benefits Enhanced Formulary PDL January
PENICILLINS ketorolac tromethamineQL GENERIC mefenamic acid amoxicillin/clavulanate potassium nabumetone amoxicillin/clavulanate potassium ER naproxen January 2016 ampicillin naproxen sodium ampicillin sodium naproxen sodium CR ESSENTIAL HEALTH BENEFITS ampicillin-sulbactam naproxen sodium ER ENHANCED PREFERRED DRUG LIST nafcillin sodium naproxen DR The Optum Preferred Drug List is a guide identifying oxacillin sodium oxaprozin preferred brand-name medicines within select penicillin G potassium piroxicam therapeutic categories. The Preferred Drug List may piperacillin sodium/ tazobactam sulindac not include all drugs covered by your prescription sodium tolmetin sodium drug benefit. Generic medicines are available within many of the therapeutic categories listed, in addition piperacillin sodium/tazobactam Fenoprofen Calcium sodium to categories not listed, and should be considered Meclofenamate Sodium piperacillin/tazobactam as the first line of prescribing. Tolmetin Sodium Amoxicillin/Clavulanate Potassium LOW COST GENERIC PREFERRED For benefit coverage or restrictions please check indomethacin your benefit plan document(s). This listing is revised Augmentin meloxicam periodically as new drugs and new prescribing LOW COST GENERIC naproxen kit information becomes available. It is recommended amoxicillin that you bring this list of medications when you or a dicloxacillin sodium CARDIOVASCULAR covered family member sees a physician or other penicillin v potassium ACE-INHIBITORS healthcare provider. GENERIC QUINOLONES captopril ANTI-INFECTIVES -

Horsemen's Information 2016和文TGP 1 薬物修正後 E
[Conditions] 1 Date December 29 (Tue), 2020 2020 Oi Racetrack, Race 10 2 Location TCK, Oi Racetrack 3 Race The 66th Running of Tokyo Daishoten (GI) 4 Eligibility Thoroughbreds, 3 years old & up 5 Full Gate 16 horses 6 Foreign Runners Selected by the selection committee from among the pre-entered horses. 7 Distance 2,000m, 1 1/4 mile (Right-handed, dirt course) 8 Weight 3 years old: 121.5 lbs,4 years old & up: 125.5 lbs, Female: 4.4 lbs less For 3 year-old-horses from the southern hemisphere, reduce 4.4 lbs from the above weight. 9 Purse Unit: 1,000 JPY Prize Purse & Bonus money Running Record 1st place 6th place allowances prize *1 prize *2 1st place 2nd place 3rd place 4th place 5th place or lower Owner 80,000 28,000 16,000 8,000 4,000 300 300 50 1,600 Trainer 880 70 60 50 40 30 30 80 Jockey 120 110 100 80 70 60 30 80 Groom 80 70 60 50 40 30 30 80 Rider 30 30 80 *1 Paid for the runner who broke the previous record and also set the best record during the race. *2 Prize equivalent to the amount listed in the table above is presented. *3 1 USD= 105.88 JPY (As of August,2020) 10 Handling of Late Scratch No allowance is paid in the case of a late scratch (including cancelation of race due to standstill in a starting gate) approved by TCK, Stewards, and Starter. However, if the chairman of the race meeting operation committee deems that the horse is involved in an accident not caused by the horse, the owner is given an running allowance. -

Survey of Pain Knowledge and Analgesia in Dogs and Cats by Colombian Veterinarians
veterinary sciences Article Survey of Pain Knowledge and Analgesia in Dogs and Cats by Colombian Veterinarians Carlos Morales-Vallecilla 1, Nicolas Ramírez 1, David Villar 1,*, Maria Camila Díaz 1 , Sandra Bustamante 1 and Duncan Ferguson 2 1 Facultad de Ciencias Agrarias Universidad de Antioquia, Medellín 050010, Colombia; [email protected] (C.M.-V.); [email protected] (N.R.); [email protected] (M.C.D.); [email protected] (S.B.) 2 Department of Comparative Biosciences, College of Veterinary Medicine, University of Illinois at Urbana-Champaign, Urbana, IL 61802, USA; [email protected] * Correspondence: [email protected]; Tel.: +57-3178047381 Received: 6 December 2018; Accepted: 5 January 2019; Published: 10 January 2019 Abstract: A questionnaire study was conducted among 131 veterinarians practicing in the city of Medellin, Colombia, to assess views on pain evaluation and management in dogs and cats. When pain recognition and quantification abilities were used as a perceived competence of proper pain assessment, only 83/131 (63.4%, confidence interval (CI) 0.55–0.72) were deemed to have satisfactory skills, with the rest considered to be deficient. There were 49/131 (37.4) veterinarians who had participated in continuing education programs and were more confident assessing pain, with an odds ratio ( standard error) of 2.84 1.15 (p = 0.01; CI 1.27–6.32). In addition, the odds of using ± ± pain scales was 4.28 2.17 (p < 0.01, CI 1.58–11.55) greater if they had also participated in continuing ± education programs. The term multimodal analgesia was familiar to 77 (58.7%) veterinarians who also claimed to use more than one approach to pain control. -

Yellox, Bromfenac
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Yellox 0.9 mg/ml eye drops, solution 2. QUALITATIVE AND QUANTITATIVE COMPOSITION One ml of solution contains 0.9 mg bromfenac (as sodium sesquihydrate). One drop contains approximately 33 micrograms bromfenac. Excipient: Each ml of solution contains 50 micrograms of benzalkonium chloride. For a full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Eye drops, solution. Clear yellow solution. pH: 8.1-8.5; osmolality: 270-330 mOsmol/kg 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Treatment of postoperative ocular inflammation following cataract extraction in adults. 4.2 Posology and method of administration Posology Use in adults, including the elderly The dose is one drop of Yellox in the affected eye(s) twice daily, beginning the next day after cataract surgery and continuing through the first 2 weeks of the postoperative period. The treatment should not exceed 2 weeks as safety data beyond this is not available. Paediatric population The safety and efficacy of bromfenac in paediatric patients has not been established. No data are available. Hepatic and renal impairment Yellox has not been studied in patients with hepatic disease or renal impairment. Method of administration For ocular use. If more than one topical ophtalmic medicinal product is being used, each one should be administered at least 5 minutes apart. To prevent contamination of the dropper-tip and solution, care must be taken not to touch the eyelids, surrounding areas or other surfaces with the dropper-tip of the bottle. Instruct patient to keep the bottle tightly closed when not in use. -
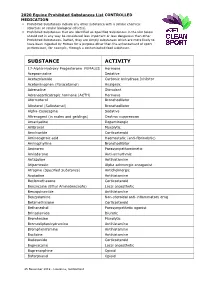
2020 Equine Prohibited Substances List CONTROLLED MEDICATION
2020 Equine Prohibited Substances List CONTROLLED MEDICATION . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). Prohibited Substances that are identified as Specified Substances in the List below should not in any way be considered less important or less dangerous than other Prohibited Substances. Rather, they are simply substances which are more likely to have been ingested by Horses for a purpose other than the enhancement of sport performance, for example, through a contaminated food substance. SUBSTANCE ACTIVITY 17-Alpha-Hydroxy Progesterone FEMALES Hormone Acepromazine Sedative Acetazolamide Carbonic Anhydrase Inhibitor Acetominophen (Paracetamol) Analgesic Adrenaline Stimulant Adrenocorticotropic hormone (ACTH) Hormone Aformoterol Bronchodilator Albuterol (Salbutamol) Bronchodilator Alpha-Casozepine Sedative Altrenogest (in males and geldings) Oestrus suppression Amantadine Dopaminergic Ambroxol Mucolytic Amcinonide Corticosteroid Aminocaproic acid Haemostatic (anti-fibrinolytic) Aminophylline Bronchodilator Aminorex Parasympathomimetic Amiodarone Anti-arrhythmic Antazoline Antihistamine Atipamezole Alpha adrenergic antagonist Atropine (Specified Substance) Anticholinergic Azatadine Antihistamine Beclomethasone Corticosteroid Benzocaine (Ethyl Aminobenzoate) Local anaesthetic Benzquinamide Antihistamine Benzydamine Non-steroidal anti-inflammatory drug Betamethasone Corticosteroid Bethanechol Parasympathetic agonist Brinzolamide Diuretic Bromhexine Mucolytic Bromodiphenhydramine -

An End-To-End Workflow for Quantitative Screening of Multiclass, Multiresidue Veterinary Drugs in Meat Using the Agilent 6470 Triple Quadrupole LC/MS
Application Note Food Testing & Agriculture An End-To-End Workflow for Quantitative Screening of Multiclass, Multiresidue Veterinary Drugs in Meat Using the Agilent 6470 Triple Quadrupole LC/MS Authors Abstract Siji Joseph, Aimei Zou, Chee A comprehensive LC/MS/MS workflow was developed for targeted screening or Sian Gan, Limian Zhao, and quantitation of 210 veterinary drug residues in animal muscle prepared for human Patrick Batoon consumption, with the intention to accelerate and simplify routine laboratory testing. Agilent Technologies, Inc. The workflow ranged from sample preparation through chromatographic separation, MS detection, data processing and analysis, and report generation. The workflow performance was evaluated using three muscle matrices—chicken, pork, and beef— and was assessed on two different Agilent triple quadrupole LC/MS models (an Agilent 6470 and a 6495C triple quadrupole LC/MS). A simple sample preparation protocol using Agilent Captiva EMR—Lipid cartridges provided efficient extraction and matrix cleanup. A single chromatographic method using Agilent InfinityLab Poroshell 120 EC-C18 columns with a 13-minute method delivered acceptable separation and retention time distribution across the elution window for reliable triple quadrupole detection and data analysis. Workflow performance was evaluated based on evaluation of limit of detection (LOD), limit of quantitation (LOQ), calibration curve linearity, accuracy, precision, and recovery, using matrix-matched spike samples for a range from 0.1 to 100 μg/L. Calibration curves were plotted from LOQ to 100 μg/L, where all analytes demonstrated linearity R2 >0.99. Instrument method accuracy values were within 73 to 113%. Target analytes response and retention time %RSD values were ≤19% and ≤0.28% respectively. -

Assessment of the Efficacy of Firocoxib and Robenacoxib
Assessment of the Efficacy of Firocoxib and Robenacoxib in an Induced Synovitis Model of Acute Arthritis in Dogs Christelle Dauteloupa Corinne Pichoub Frederic Beugneta,* aMerial SAS, 2 Av Pasteur, 69007, Lyon, France b Amatsigroup, Site AmatsiAvogadro, Parc de Génibrat, 31470 Fontenilles, France * Corresponding author: E-mail address: [email protected] KEY WORDS: Firocoxib, Robenacoxib, and the increased PVF values compared to Arthritis, Lameness score, Peak Vertical the control group at 3 and 5 hours post- force injection. Firocoxib performed significantly better than robenacoxib at 3, 5, and 10 hours ABSTRACT post-UC injection. In this model, robena- The objective of this study was to compare coxib was not different from control for both the analgesic activity of a single oral dose the VLS and the PVF values. Pre-treatment of two COX-2 selective inhibitors, firo- with firocoxib reduced the induced pain as- coxib (Previcox®, Merial) and robenacoxib sociated with intra-articular administration (Onsior®, Elanco), in an acute pain model of urate crystals. in dogs. Sixteen healthy Beagle dogs were randomly allocated to three groups. Two INTRODUCTION successive experiments were conducted, in Chronic osteoarthritis is common in dogs which eight dogs served as control. Eight and is estimated to affect 20% of dogs dogs received firocoxib or robenacoxib in a over 1 year of age (Johnston, 1997). Since cross-over design at the recommended dos- no drug has been shown to reverse the age 13 hours before intra-articular injection pathological changes of osteoarthritis, the of a urate crystal suspension (UC) for induc- objective of treatment is to reduce pain and tion of synovitis. -

NIH Public Access Author Manuscript J Am Chem Soc
NIH Public Access Author Manuscript J Am Chem Soc. Author manuscript; available in PMC 2014 January 09. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: J Am Chem Soc. 2013 January 9; 135(1): 22–25. doi:10.1021/ja308733u. A binding site for non-steroidal anti-inflammatory drugs in FAAH Laura Bertolacci¶, Elisa Romeo¶, Marina Veronesi, Paola Magotti, Clara Albani, Mauro Dionisi, Chiara Lambruschini, Rita Scarpelli, Andrea Cavalli‡, Marco De Vivo, Daniele Piomelli*,†, and Gianpiero Garau* ‡Department of Pharmaceutical. Sciences, University of Bologna, Italy. †Department of Pharmacology, Univ. of California, Irvine, California, USA. Abstract In addition to inhibiting the cyclooxygenasemediated biosynthesis of prostanoids, various widely used non-steroidal anti-inflammatory drugs (NSAIDs) enhance endocannabinoid signaling by blocking the anandamidedegrading membrane enzyme, fatty acid amide hydrolase (FAAH). The X-ray structure of FAAH in complex with the NSAID carprofen, along with studies of site- directed mutagenesis, enzyme activity assays, and nuclear magnetic resonance, now reveal the molecular details of this interaction, providing information that may guide the design of dual FAAH-cyclooxygenase inhibitors with superior analgesic efficacy. Non-steroidal anti-inflammatory drugs (NSAIDs), one of the most widely used classes of therapeutic agents, alleviate pain and inflammation1 by inhibiting the enzymes cyclooxygenase-1 (COX-1) and COX-2,2 which catalyze the conversion of -

Use of Topical Bromfenac for Treating Ocular Pain and Inflammation Beyond Cataract Surgery: a Review of Published Studies
Clinical Ophthalmology Dovepress open access to scientific and medical research Open Access Full Text Article REVIEW Use of topical bromfenac for treating ocular pain and inflammation beyond cataract surgery: a review of published studies This article was published in the following Dove Press journal: Clinical Ophthalmology Barry A Schechter Abstract: Topical ophthalmic nonsteroidal anti-inflammatory drugs (NSAIDs) are com- monly used to treat postoperative inflammation and pain following cataract surgery and for Cornea and Cataract Service, Florida Eye Microsurgical Institute, Boynton Beach, treatment and prophylaxis of pseudophakic cystoid macular edema (CME). Bromfenac is a FL, USA brominated NSAID with strong in vitro anti-inflammatory potency. Like other ophthalmic NSAIDs, bromfenac is often used outside of the cataract surgery setting. This paper provides an overview of bromfenac’s preclinical ocular pharmacology and pharmacokinetics, followed by a review of 23 published clinical studies in which various marketed bromfenac formula- tions were used for conditions other than cataract surgery or pseudophakic CME. These include: post-refractive eye surgery; macular edema associated with diabetes, uveitis, or fl For personal use only. retinal vein occlusion; in ammation associated with age-related macular degeneration; pain related to intravitreal injections; and other ocular anterior segment and surface disorders with an inflammatory component. The published evidence reviewed supports the safety and effectiveness of bromfenac in these additional ophthalmic indications. Bromfenac was well tolerated when given alone or in combination with intravitreal anti-vascular endothelial growth factor agents, topical corticosteroids, or topical mast-cell stabilizers. The most common adverse event reported was ocular irritation. No serious adverse events (ie, corneal epithelial disorders) were reported, although the majority of studies did not systematically evaluate potential side effects. -

A. List of Prohibited Substances 1
Reviewed 09.05.17 List over prohibited substances and withdrawal times in Scandinavia, valid from May 20th, 2017 This list has been developed in collaboration with the other Scandinavian countries through NEMAC (Nordic Equine Medication and Anti-doping Committee) The List of prohibited substances and withdrawal times consists of the A-list, listing substances and treatment methods that are absolutely prohibited for horses, and the B-list, listing substances that are prohibited in competition; the withdrawal times for these substances as well as treatment methods with withdrawal times. The list may be reviewed several times per year. This list is valid starting from May 20th, 2017 and is enforced until a new list takes effect. A valid list of withdrawal times can also be found at any time on DNT’s official website, www.travsport.no.ST’s official website, www.travsport.se, and DTC`s official website www.trav.dk Health certificate and keeping of medical records The trainer is responsible for ensuring that any treatment that requires a withdrawal time is listed in the horse’s health certificate. The start and end dates of the treatment, the name of the treatment/medication/active substance, amount given, method of administration, withdrawal time as well as the name of the veterinarian or other person responsible for the treatment must all be listed in the health certificate in accordance with DNT’s Doping Regulations 2017 § 4. Passport and health certificate should be brought with the horse at all times. Omitting, incompletely or improperly listing treatments in the health certificate constitutes a breach of the Doping Regulations. -

(12) United States Patent (10) Patent No.: US 6,472,433 B2 Wechter (45) Date of Patent: Oct
USOO6472433B2 (12) United States Patent (10) Patent No.: US 6,472,433 B2 Wechter (45) Date of Patent: Oct. 29, 2002 (54) METHOD FOR TREATMENT OF Variability of Inversion of (R)-Flurbiprofen in Different NFLAMMATION WITH R-NSAIDS Species, Sabine Menzel-Soglowek, Gerd Geisslinger, Win fried S. Beck, and Kay Brune-Journal of Pharmaceutical (75) Inventor: William J. Wechter, Ojai, CA (US) Sciences vol. 81, No. 9, Sep. 1992. Disposition and Pharmacokinetics of R-flurbiprofen in (73) Assignee: Loma Linda University Medical Three Species: Demonstration of R- to S-Flurbiprofen Center, Loma Linda, CA (US) Inversion in the Mouse, Rat and Monkey-William J. Wechter, E. David Murray, Jr. Karina M. Gibson, David D. (*) Notice: Subject to any disclaimer, the term of this Quiggle, and Douglas L. Leipold-Laboratory of Chemical patent is extended or adjusted under 35 Endocrinology, Loma Linda University School of Medicine, U.S.C. 154(b) by 0 days. 1998. Superaspirin, Jerome Groopman The New Yorker, Jun. 15, (21) Appl. No.: 09/797,022 1998 pp. 32–35. (22) Filed: Mar. 1, 2001 Building a Better Aspirin, Science, vol. 280, May 22, 1998. (65) Prior Publication Data R-Flurbiprofen Chemoprevention and Treatment of Intesti nal Adenomas in the APC Min/+ Mouse Model: Implica US 2001/0012849 A1 Aug. 9, 2001 tions for Prophylazis and Treatment of Colon Caner, Will iam Wechter, Darko Kantoci, E. David Murray, Jr. David D. Related U.S. Application Data Quiggle, Douglas D. Leipold, Karina M. Gibson, and John D. McCracker-Cancer Research 57, 4316-4324, Oct. 1, (63) Continuation of application No.