Utilization of Formate in Two Biochemical Reactions in Gram-Negative Bacteria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

B Number Gene Name Mrna Intensity Mrna Present # of Tryptic
list list sample) short list predicted B number Gene name assignment mRNA present mRNA intensity Gene description Protein detected - Membrane protein detected (total list) detected (long list) membrane sample Proteins detected - detected (short list) # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides Functional category detected (membrane Protein detected - total Protein detected - long b0003 thrB 6781 P 9 P 3 3 P 3 0 homoserine kinase Metabolism of small molecules b0004 thrC 15039 P 18 P 10 P 11 P 10 0 threonine synthase Metabolism of small molecules b0008 talB 20561 P 20 P 13 P 16 P 13 0 transaldolase B Metabolism of small molecules b0009 mog 1296 P 7 0 0 0 0 required for the efficient incorporation of molybdate into molybdoproteins Metabolism of small molecules b0014 dnaK 13283 P 32 P 23 P 24 P 23 0 chaperone Hsp70; DNA biosynthesis; autoregulated heat shock proteins Cell processes b0031 dapB 2348 P 16 P 3 3 P 3 0 dihydrodipicolinate reductase Metabolism of small molecules b0032 carA 9312 P 14 P 8 P 8 P 8 0 carbamoyl-phosphate synthetase, glutamine (small) subunit Metabolism of small molecules b0048 folA 1588 P 7 P 1 2 P 1 0 dihydrofolate reductase type I; trimethoprim resistance Metabolism of small molecules peptidyl-prolyl cis-trans isomerase (PPIase), involved in maturation of outer b0053 surA 3825 P 19 P 4 P 5 P 4 P(m) 1 GenProt membrane proteins (1st module) Cell processes b0054 imp 2737 P 42 P 5 0 0 P(m) 5 GenProt organic solvent tolerance Cell processes b0071 leuD 4770 -

B Number Gene Name Mrna Intensity Mrna
sample) total list predicted B number Gene name assignment mRNA present mRNA intensity Gene description Protein detected - Membrane protein membrane sample detected (total list) Proteins detected - Functional category # of tryptic peptides # of tryptic peptides # of tryptic peptides detected (membrane b0002 thrA 13624 P 39 P 18 P(m) 2 aspartokinase I, homoserine dehydrogenase I Metabolism of small molecules b0003 thrB 6781 P 9 P 3 0 homoserine kinase Metabolism of small molecules b0004 thrC 15039 P 18 P 10 0 threonine synthase Metabolism of small molecules b0008 talB 20561 P 20 P 13 0 transaldolase B Metabolism of small molecules chaperone Hsp70; DNA biosynthesis; autoregulated heat shock b0014 dnaK 13283 P 32 P 23 0 proteins Cell processes b0015 dnaJ 4492 P 13 P 4 P(m) 1 chaperone with DnaK; heat shock protein Cell processes b0029 lytB 1331 P 16 P 2 0 control of stringent response; involved in penicillin tolerance Global functions b0032 carA 9312 P 14 P 8 0 carbamoyl-phosphate synthetase, glutamine (small) subunit Metabolism of small molecules b0033 carB 7656 P 48 P 17 0 carbamoyl-phosphate synthase large subunit Metabolism of small molecules b0048 folA 1588 P 7 P 1 0 dihydrofolate reductase type I; trimethoprim resistance Metabolism of small molecules peptidyl-prolyl cis-trans isomerase (PPIase), involved in maturation of b0053 surA 3825 P 19 P 4 P(m) 1 GenProt outer membrane proteins (1st module) Cell processes b0054 imp 2737 P 42 P 5 P(m) 5 GenProt organic solvent tolerance Cell processes b0071 leuD 4770 P 10 P 9 0 isopropylmalate -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

(10) Patent No.: US 8119385 B2
US008119385B2 (12) United States Patent (10) Patent No.: US 8,119,385 B2 Mathur et al. (45) Date of Patent: Feb. 21, 2012 (54) NUCLEICACIDS AND PROTEINS AND (52) U.S. Cl. ........................................ 435/212:530/350 METHODS FOR MAKING AND USING THEMI (58) Field of Classification Search ........................ None (75) Inventors: Eric J. Mathur, San Diego, CA (US); See application file for complete search history. Cathy Chang, San Diego, CA (US) (56) References Cited (73) Assignee: BP Corporation North America Inc., Houston, TX (US) OTHER PUBLICATIONS c Mount, Bioinformatics, Cold Spring Harbor Press, Cold Spring Har (*) Notice: Subject to any disclaimer, the term of this bor New York, 2001, pp. 382-393.* patent is extended or adjusted under 35 Spencer et al., “Whole-Genome Sequence Variation among Multiple U.S.C. 154(b) by 689 days. Isolates of Pseudomonas aeruginosa” J. Bacteriol. (2003) 185: 1316 1325. (21) Appl. No.: 11/817,403 Database Sequence GenBank Accession No. BZ569932 Dec. 17. 1-1. 2002. (22) PCT Fled: Mar. 3, 2006 Omiecinski et al., “Epoxide Hydrolase-Polymorphism and role in (86). PCT No.: PCT/US2OO6/OOT642 toxicology” Toxicol. Lett. (2000) 1.12: 365-370. S371 (c)(1), * cited by examiner (2), (4) Date: May 7, 2008 Primary Examiner — James Martinell (87) PCT Pub. No.: WO2006/096527 (74) Attorney, Agent, or Firm — Kalim S. Fuzail PCT Pub. Date: Sep. 14, 2006 (57) ABSTRACT (65) Prior Publication Data The invention provides polypeptides, including enzymes, structural proteins and binding proteins, polynucleotides US 201O/OO11456A1 Jan. 14, 2010 encoding these polypeptides, and methods of making and using these polynucleotides and polypeptides. -

Product Sheet Info
Master Clone List for NR-19274 Mycobacterium tuberculosis Gateway® Clone Set, Recombinant in Escherichia coli, Plates 1-42 Catalog No. NR-19274 Table 1: Mycobacterium tuberculosis, Gateway® Clones, Plate 1 (ZMTDA), NR-19637 Clone Well ORF Locus ID Description (Gene name) Accession Average Depth Position Length Number of Coverage 71201 A01 124 Rv1572c hypothetical protein Rv1572c NP_216088.2 2 71005 A02 151 Rv3461c 50S ribosomal protein L36 (rpmJ) NP_217978.1 2 71053 A03 181 Rv3924c 50S ribosomal protein L34 (rpmH) 2 71013 A04 184 Rv2452c hypothetical protein Rv2452c NP_216968.1 2 71167 A05 193 Rv0657c hypothetical protein Rv0657c NP_215171.1 2.69948187 71177 A06 211 Rv0666 hypothetical protein Rv0666 NP_215180.1 2 71225 A07 214 Rv1693 hypothetical protein Rv1693 NP_216209.1 2 71073 A08 217 Rv2099c PE family protein (PE21) 2 70874 A09 220 Rv0810c hypothetical protein Rv0810c NP_215325.1 2 70913 A10 223 Rv2371 PE-PGRS family protein (PE_PGRS40) YP_177875.1 2 71141 A11 229 Rv2806 hypothetical protein Rv2806 NP_217322.1 2 71121 A12 235 Rv1113 hypothetical protein Rv1113 NP_215629.1 1.99574468 71181 B01 241 Rv3648c cold shock protein A (cspA) NP_218165.1 2 70937 B02 244 Rv0763c ferredoxin NP_215277.1 2 70966 B03 247 Rv1054 integrase NP_215570.2 1.27530364 71145 B04 253 Rv2377c putative protein MbtH (mbtH) NP_216893.1 2 70861 B05 253 Rv2830c hypothetical protein Rv2830c NP_217346.1 2 70853 B06 253 Rv3221c anti-sigma factor YP_177945.1 2 71210 B07 256 Rv1893 hypothetical protein Rv1893 NP_216409.1 2 71062 B08 259 Rv0378 glycine rich protein -

Product Sheet Info
Master Clone List for NR-19279 ® Vibrio cholerae Gateway Clone Set, Recombinant in Escherichia coli, Plates 1-46 Catalog No. NR-19279 Table 1: Vibrio cholerae Gateway® Clones, Plate 1 (NR-19679) Clone ID Well ORF Locus ID Symbol Product Accession Position Length Number 174071 A02 367 VC2271 ribD riboflavin-specific deaminase NP_231902.1 174346 A03 336 VC1877 lpxK tetraacyldisaccharide 4`-kinase NP_231511.1 174354 A04 342 VC0953 holA DNA polymerase III, delta subunit NP_230600.1 174115 A05 388 VC2085 sucC succinyl-CoA synthase, beta subunit NP_231717.1 174310 A06 506 VC2400 murC UDP-N-acetylmuramate--alanine ligase NP_232030.1 174523 A07 132 VC0644 rbfA ribosome-binding factor A NP_230293.2 174632 A08 322 VC0681 ribF riboflavin kinase-FMN adenylyltransferase NP_230330.1 174930 A09 433 VC0720 phoR histidine protein kinase PhoR NP_230369.1 174953 A10 206 VC1178 conserved hypothetical protein NP_230823.1 174976 A11 213 VC2358 hypothetical protein NP_231988.1 174898 A12 369 VC0154 trmA tRNA (uracil-5-)-methyltransferase NP_229811.1 174059 B01 73 VC2098 hypothetical protein NP_231730.1 174075 B02 82 VC0561 rpsP ribosomal protein S16 NP_230212.1 174087 B03 378 VC1843 cydB-1 cytochrome d ubiquinol oxidase, subunit II NP_231477.1 174099 B04 383 VC1798 eha eha protein NP_231433.1 174294 B05 494 VC0763 GTP-binding protein NP_230412.1 174311 B06 314 VC2183 prsA ribose-phosphate pyrophosphokinase NP_231814.1 174603 B07 108 VC0675 thyA thymidylate synthase NP_230324.1 174474 B08 466 VC1297 asnS asparaginyl-tRNA synthetase NP_230942.2 174933 B09 198 -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Vitamin Biosynthesis As an Antifungal Target
Journal of Fungi Review Vitamin Biosynthesis as an Antifungal Target Zohar Meir and Nir Osherov * Department of Clinical Microbiology and Immunology, Sackler School of Medicine, Tel-Aviv University, Ramat-Aviv, Tel-Aviv 69978, Israel; [email protected] * Correspondence: [email protected]; Tel.: +972-3-640-9599; Fax: +972-3-640-9160 Received: 29 May 2018; Accepted: 13 June 2018; Published: 17 June 2018 Abstract: The large increase in the population of immunosuppressed patients, coupled with the limited efficacy of existing antifungals and rising resistance toward them, have dramatically highlighted the need to develop novel drugs for the treatment of invasive fungal infections. An attractive possibility is the identification of possible drug targets within essential fungal metabolic pathways not shared with humans. Here, we review the vitamin biosynthetic pathways (vitamins A–E, K) as candidates for the development of antifungals. We present a set of ranking criteria that identify the vitamin B2 (riboflavin), B5 (pantothenic acid), and B9 (folate) biosynthesis pathways as being particularly rich in new antifungal targets. We propose that recent scientific advances in the fields of drug design and fungal genomics have developed sufficiently to merit a renewed look at these pathways as promising sources for the development of novel classes of antifungals. Keywords: antifungals; fungal vitamin metabolism; drug target; essential genes 1. Introduction The number of life-threatening fungal infections has risen dramatically over the last twenty years. Recent estimates have identified a global burden of almost two million patients with systemic and invasive fungal infections, including ~700,000 cases of invasive candidiasis, ~500,000 cases of Pneumocystis jirovecii pneumonia, ~250,000 cases of invasive aspergillosis, ~220,000 cases of cryptococcal meningitis, and ~100,000 cases of disseminated histoplasmosis [1,2]. -

Investigation of the Binding of Epimer a of the Covalent Hydrate of 6,7-Bis
J. Org. Chem. 2002, 67, 2087-2092 2087 Investigation of the Binding of Epimer A of the Covalent Hydrate of 6,7-Bis(trifluoromethyl)-8-D-ribityllumazine to a Recombinant F22W Bacillus subtilis Lumazine Synthase Mutant by 15N{19F} REDOR NMR Anil K. Mehta,† Daniel R. Studelska,† Markus Fischer,‡ Andreas Giessauf,‡ Kristina Kemter,‡ Adelbert Bacher,‡ Mark Cushman,*,§ and Jacob Schaefer*,† Department of Chemistry, Washington University, Saint Louis, Missouri 63130, Lehrstuhl fu¨ r Organische Chemie und Biochemie, Technische Universita¨tMu¨ nchen, D-85747 Garching, Germany, and Department of Medicinal Chemistry and Molecular Pharmacology, School of Pharmacy, Purdue University, West Lafayette, Indiana 47907 [email protected]. Received September 12, 2001 The two epimeric covalent hydrates A and B of 6,7-bis(trifluoromethyl)-8-D-ribityllumazine are metabolically stable analogues of hypothetical intermediates proposed in the reactions catalyzed by riboflavin synthase and lumazine synthase. To confirm the stereochemical assignments previously based solely on results for epimer B, a 15N{19F} REDOR NMR study was performed on the complex formed from epimer A and a recombinant, uniformly 15N-labeled F22W mutant of Bacillus subtilis lumazine synthase. The results indicate that the fluorines of the ligands are closer to the side chain nitrogens of Arg127 and farther away from the side chain nitrogens of Lys135 in epimer B than in epimer A. These results are consistent with the assignment of the earlier 7R configuration of epimer A and the 7S configuration of epimer B. Riboflavin synthase catalyzes a mechanistically com- Scheme 1 plex and incompletely understood dismutation reaction involving the transfer of a four-carbon unit from one molecule of 6,7-dimethyl-8-D-ribityllumazine (3)toan- other molecule of 3, resulting in the formation of one molecule of riboflavin (4) and one molecule of the ribi- tylaminopyrimidine 1 (Scheme 1). -
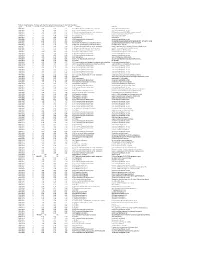
087643-10.Pdf
Table S4. Gene Expression For Genes with Top 50% Expression Values during 12 Hours Post Starvation Name Number log2 Fold Change Standard Deviation Standard Error p-value Category Definition PMM0001 2 0.89 1.01 0.71 0.54 DNA replication, recombination, and repair DNA polymerase III, beta chain PMM0008 2 1.34 0.16 0.11 0.28 Conserved hypothetical protein conserved hypothetical protein PMM0013 2 -1.48 0.60 0.42 0.88 Fatty acid, phospholipid and sterol metabolism RNA-binding region RNP-1 (RNA recognition motif) PMM0015 2 0.25 0.37 0.26 0.93 Conserved hypothetical protein Domain of unknown function DUF25 PMM0016 2 -1.51 1.08 0.76 0.27 Chaperones Heat shock protein GrpE PMM0017 2 1.71 0.02 0.01 0.10 Chaperones DnaJ protein PMM0020 2 -0.90 2.70 1.91 1.00 Conserved hypothetical protein conserved hypothetical protein PMM0023 2 -0.58 0.75 0.53 0.46 CO2 fixation Glyceraldehyde 3-phosphate dehydrogenase(NADP+)(phosphorylating) PMM0025 2 0.18 0.31 0.22 0.83 Protein modification and translation factors Cyclophilin-type peptidyl-prolyl cis-trans isomerase PMM0026 2 -1.42 1.65 1.16 0.08 Protein modification and translation factors Elongation factor P (EF-P) PMM0027 2 0.53 0.68 0.48 1.00 Fatty acid, phospholipid and sterol metabolism Biotin / Lipoyl attachment:Acetyl-CoA biotin carboxyl carrier... PMM0030 2 5.12 1.40 0.99 0.00 Protein modification and translation factors possible Transcription factor TFIID (or TATA-b PMM0031 2 1.08 0.16 0.11 0.87 DNA replication, recombination, and repair HNH endonuclease:HNH nuclease PMM0032 2 0.40 0.91 0.65 0.82 Protein -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000190595Cyc: Cellulophaga lytica DSM 7489 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari phosphate molybdate RS04350 RS08315 phosphate molybdate Macromolecule Modification tRNA-uridine 2-thiolation inosine 5'- Nucleoside and Nucleotide Degradation ditrans,octacis- a peptidoglycan with ditrans,octacis- a [bis(guanylyl an N-terminal- a [protein] and selenation (bacteria) phosphate guanosine ditrans,octacis- (4R)-4-hydroxy- an L-asparaginyl- an L-cysteinyl- Aminoacyl-tRNA Charging tRNA charging nucleotides undecaprenyldiphospho- (L-alanyl-γ-D-glutamyl- undecaprenyldiphospho- undecaprenyldiphospho- molybdopterin) cys L-methionyl-L- C-terminal glu degradation 2-oxoglutarate [tRNA Asn ] [tRNA Cys ] degradation III N-acetyl-(N-acetyl-β-D- L-lysyl-D-alanyl-D- N-acetyl-(N- N-acetyl-(N- cofactor cysteinyl-[protein] L-glutamate cys glucosaminyl)muramoyl- alanine) pentapeptide acetylglucosaminyl) acetylglucosaminyl) chaperone]- Phe IMP bifunctional phe a tRNA L-alanyl-γ-D-glutamyl-L- -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine