Diplomarbeit
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
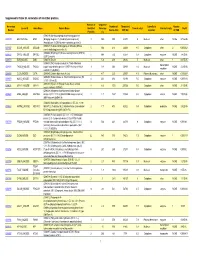
Supporting Table S3 for PDF Maker
Supplemental Table S3. Annotation of identified proteins. Number of Sequence Accession Number of Theoretical Subcellular Number Locus ID Gene Name Protein Name Identified Coverage Theoretical pI Protein Family NSAF Number Amino Acid MW (Da) Location of TMD Peptides (%) (O08539) Myc box-dependent-interacting protein 1 O08539 BIN1_MOUSE BIN1 (Bridging integrator 1) (Amphiphysin-like protein) 3 10.5 588 64470 5 Nucleus other NONE 5.73E-05 (Amphiphysin II) (SH3-domain-containing protein 9) (O08547) Vesicle-trafficking protein SEC22b (SEC22 O08547 SC22B_MOUSE SEC22B 5 30.4 214 24609 8.5 Cytoplasm other 2 0.000262 vesicle-trafficking protein-like 1) (O08553) Dihydropyrimidinase-related protein 2 (DRP-2) O08553 DPYL2_MOUSE DPYSL2 5 19.4 572 62278 6.4 Cytoplasm enzyme NONE 7.85E-05 (ULIP 2 protein) O08579 EMD_MOUSE EMD (O08579) Emerin 1 5.8 259 29436 5 Nucleus other 1 8.67E-05 (O08583) THO complex subunit 4 (Tho4) (RNA and transcription O08583 THOC4_MOUSE THOC4 export factor-binding protein 1) (REF1-I) (Ally of AML-1 1 9.8 254 26809 11.2 Nucleus NONE 2.21E-05 regulator and LEF-1) (Aly/REF) O08585 CLCA_MOUSE CLTA (O08585) Clathrin light chain A (Lca) 2 4.7 235 25557 4.5 Plasma Membrane other NONE 0.000287 (O08600) Endonuclease G, mitochondrial precursor (EC O08600 NUCG_MOUSE ENDOG 4 23.1 294 32191 9.5 Cytoplasm enzyme NONE 0.000134 3.1.30.-) (Endo G) (O08638) Myosin-11 (Myosin heavy chain, smooth O08638 MYH11_MOUSE MYH11 8 6.6 1972 227026 5.5 Cytoplasm other NONE 3.13E-05 muscle isoform) (SMMHC) (O08648) Mitogen-activated protein kinase kinase -

Coa Protects Against the Deleterious Effects of Caloric Overload in Drosophila Laura Palanker Musselman Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2016 CoA protects against the deleterious effects of caloric overload in Drosophila Laura Palanker Musselman Washington University School of Medicine in St. Louis Jill L. Fink Washington University School of Medicine in St. Louis Thomas J. Baranski Washington University School of Medicine in St. Louis Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Musselman, Laura Palanker; Fink, Jill L.; and Baranski, Thomas J., ,"CoA protects against the deleterious effects of caloric overload in Drosophila." Journal of Lipid Research.57,3. 380-387. (2016). https://digitalcommons.wustl.edu/open_access_pubs/5521 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. CoA protects against the deleterious effects of caloric overload in Drosophila 1 Laura Palanker Musselman , 2 Jill L. Fink , and Thomas J. Baranski 3 Division of Endocrinology, Metabolism, and Lipid Research, Department of Medicine, Washington University School of Medicine , St. Louis, MO 63110 Abstract We developed a Drosophila model of T2D in in various tissues ( 5–9 ). These lipid mediators induce cel- which high sugar (HS) feeding leads to insulin resistance. In lular stress known as lipid toxicity or “lipotoxicity” ( 4, 10, this model, adipose TG storage is protective against fatty 11 ). Therefore, it is of interest to learn the mechanisms Downloaded from acid toxicity and diabetes. Initial biochemical and gene ex- that enable animals to store excess carbons safely as TG in pression studies suggested that defi ciency in CoA might un- the face of caloric overload. -

Is Pantetheinase the Actual Identity of Mouse and Human Vanin-1 Proteins?
FEBS 22873 FEBS Letters 461 (1999) 149^152 View metadata, citation and similar papers at core.ac.uk brought to you by CORE Is pantetheinase the actual identity of mouse and humanprovided vanin-1 by Elsevier - Publisher Connector proteins? Bruno Marasa;*, Donatella Barraa, Silvestro Dupre©a, Giuseppina Pitarib aDipartimento di Scienze Biochimiche `A. Rossi Fanelli' and Centro di Biologia Molecolare del Consiglio Nazionale delle Ricerche Universita© La Sapienza, Piazzale Aldo Moro 5, 00185 Rome, Italy bDipartimento di Biologia di Base ed Applicata, Universita© de L'Aquila Via Vetoio-Coppito, 67100 L'Aquila, Italy Received 12 October 1999 [6]. Data on molecular weight, isoelectric point, amino acid Abstract Pantetheinase is an amidohydrolase involved in the dissimilative pathway of CoA, allowing the turnover of the and carbohydrate content and other properties of proteins of pantothenate moiety. We have determined the N-terminal di¡erent origin have been published [2,5]. Details on kinetic sequence as well as the sequences of a number of tryptic and and catalytic features of the horse kidney enzyme [2,3], on chymotryptic peptides of the protein isolated from pig kidney. inhibition patterns by disul¢des [12,13], on the stability of These sequence stretches were used as probes to search in the the molecule and the £exibility of the active site of the pig SwissProt database and significant similarities were found with a kidney enzyme [14,15] were also reported. GPI-anchored protein (mouse vanin-1, with a suggested role in The enzyme has a role in the dissimilative pathway of CoA lymphocyte migration), with two putative proteins encoded by and acyl carrier proteins, allowing the turnover of the pan- human cDNAs (VNN1 and VNN2) and with human biotinidase. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

Generate Metabolic Map Poster
Authors: Kenneth L Wiley, Harvard University An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Andy Schroeder, FlyBase/Harvard University Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. FlyCyc: Drosophila melanogaster Cellular Overview Connections between pathways are omitted for legibility. -

TICK-FLAVIVIRUS INTERACTIONS: DISCOVERY of HOST PROTEINS THAT ARE AFFECTED in VIRUS INFECTION Jeffrey Michael Grabowski Purdue University
Purdue University Purdue e-Pubs Open Access Dissertations Theses and Dissertations January 2015 TICK-FLAVIVIRUS INTERACTIONS: DISCOVERY OF HOST PROTEINS THAT ARE AFFECTED IN VIRUS INFECTION Jeffrey Michael Grabowski Purdue University Follow this and additional works at: https://docs.lib.purdue.edu/open_access_dissertations Recommended Citation Grabowski, Jeffrey Michael, "TICK-FLAVIVIRUS INTERACTIONS: DISCOVERY OF HOST PROTEINS THAT ARE AFFECTED IN VIRUS INFECTION" (2015). Open Access Dissertations. 1457. https://docs.lib.purdue.edu/open_access_dissertations/1457 This document has been made available through Purdue e-Pubs, a service of the Purdue University Libraries. Please contact [email protected] for additional information. TICK-FLAVIVIRUS INTERACTIONS: DISCOVERY OF HOST PROTEINS THAT ARE AFFECTED IN VIRUS INFECTION A Dissertation Submitted to the Faculty of Purdue University by Jeffrey Michael Grabowski In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy August 2015 Purdue University West Lafayette, Indiana ii This dissertation is dedicated to my family and friends. I thank Edgar and Mary Grabowski for the utmost support over the years. Also, Jessica and Kevin Napadow have provided great opportunities to help ease one’s mind in stressful times. In addition, I commend my aunts, uncles (present and past), and cousins for the stimulating times. Last, I have so much appreciation for Anastasia Richards and my past grandparents for the sacrifices they made to provide a foundation for the family present today. All in all, words cannot describe the love and the appreciation that I have for my family’s support and sacrifice over the years. Friendship goes a long way, and there are too many friends to list that I owe my appreciation too during these years here at Purdue University. -

Springer Handbook of Enzymes
Dietmar Schomburg Ida Schomburg (Eds.) Springer Handbook of Enzymes Alphabetical Name Index 1 23 © Springer-Verlag Berlin Heidelberg New York 2010 This work is subject to copyright. All rights reserved, whether in whole or part of the material con- cerned, specifically the right of translation, printing and reprinting, reproduction and storage in data- bases. The publisher cannot assume any legal responsibility for given data. Commercial distribution is only permitted with the publishers written consent. Springer Handbook of Enzymes, Vols. 1–39 + Supplements 1–7, Name Index 2.4.1.60 abequosyltransferase, Vol. 31, p. 468 2.7.1.157 N-acetylgalactosamine kinase, Vol. S2, p. 268 4.2.3.18 abietadiene synthase, Vol. S7,p.276 3.1.6.12 N-acetylgalactosamine-4-sulfatase, Vol. 11, p. 300 1.14.13.93 (+)-abscisic acid 8’-hydroxylase, Vol. S1, p. 602 3.1.6.4 N-acetylgalactosamine-6-sulfatase, Vol. 11, p. 267 1.2.3.14 abscisic-aldehyde oxidase, Vol. S1, p. 176 3.2.1.49 a-N-acetylgalactosaminidase, Vol. 13,p.10 1.2.1.10 acetaldehyde dehydrogenase (acetylating), Vol. 20, 3.2.1.53 b-N-acetylgalactosaminidase, Vol. 13,p.91 p. 115 2.4.99.3 a-N-acetylgalactosaminide a-2,6-sialyltransferase, 3.5.1.63 4-acetamidobutyrate deacetylase, Vol. 14,p.528 Vol. 33,p.335 3.5.1.51 4-acetamidobutyryl-CoA deacetylase, Vol. 14, 2.4.1.147 acetylgalactosaminyl-O-glycosyl-glycoprotein b- p. 482 1,3-N-acetylglucosaminyltransferase, Vol. 32, 3.5.1.29 2-(acetamidomethylene)succinate hydrolase, p. 287 Vol. -

Molecular Characterisation of Two Ornithodoros Savignyi Enzyme Isoforms from the 5
Gene expression profiling of adult female tissues in feeding Rhipicephalus microplus cattle ticks Christian Stutzera, Willem A. van Zyla, Nicholas A. Olivierb, Sabine Richardsc, Christine Maritz-Olivierc,* aDepartment of Biochemistry, Faculty of Natural and Agricultural Sciences, University of Pretoria, South Africa, 0002. bACGT DNA microarray facility, Department of Plant Sciences, Faculty of Natural and Agricultural Sciences, University of Pretoria, South Africa, 0002. cDepartment of Genetics, Faculty of Natural and Agricultural Sciences, University of Pretoria, South Africa, 0002. *Corresponding author:.Tel: +27 012 420 3945; fax: +27 012 362 5327 E-mail address: [email protected] Note: Supplementary data associated with this article. Note: The microarray and sequence data related to this work is available via the NCBI GEO database, GEO accession no. GSE35867. 1 Abstract The southern cattle tick, Rhipicephalus microplus, is an economically important pest, especially for resource-poor countries, both as a highly adaptive invasive species and prominent vector of disease. The increasing prevalence of resistance to chemical acaricides and variable efficacy of current tick vaccine candidates highlight the need for more effective control methods. In the absence of a fully annotated genome, the wealth of available expressed sequence tag (EST) sequence data for this species presents a unique opportunity to study the genes that are expressed in tissues involved in blood meal acquisition, digestion and reproduction during feeding. Utilizing a custom oligonucleotide microarray designed from available singletons (BmiGI Version 2.1) and EST sequences of R. microplus, the expression profiles in feeding adult female midgut, salivary glands and ovarian tissues were compared. From 13,456 assembled transcripts, 588 genes expressed in all three tissues were identified from fed adult females 20 days post infestation. -

Insights Into Cis-Autoproteolysis Reveal a Reactive State Formed Through Conformational Rearrangement
Insights into cis-autoproteolysis reveal a reactive state formed through conformational rearrangement Andrew R. Bullera, Michael F. Freemanb,1, Nathan T. Wrightc,2, Joel F. Schildbachc, and Craig A. Townsendb,3 aDepartments of Biophysics, bChemistry, and cBiology, Johns Hopkins University, 3400 North Charles Street, Baltimore MD 21218 Edited by Gregory A. Petsko, Brandeis University, Waltham, MA, and approved December 20, 2011 (received for review August 19, 2011) ThnT is a pantetheine hydrolase from the DmpA/OAT superfamily for the 15-kDa Muc1 SEA domain, where it was shown that involved in the biosynthesis of the β-lactam antibiotic thienamycin. protein folding introduces torsional strain at the scissile bond, We performed a structural and mechanistic investigation into the lowering the barrier to cleavage by 7 kcal∕mol (11, 12). The Ntn cis-autoproteolytic activation of ThnT, a process that has not pre- hydrolases were the first structurally characterized cis-autopro- viously been subject to analysis within this superfamily of enzymes. teolytic enzymes (13), but their size and complexity hinder similar Removal of the γ-methyl of the threonine nucleophile resulted in a progress. Enzymes from this superfamily are known to utilize all rate deceleration that we attribute to a reduction in the population three possible nucleophiles (Thr, Ser, or Cys); however, the of the reactive rotamer. This phenomenon is broadly applicable and mechanistic constraints imparted by a given residue are entirely constitutes a rationale for the evolutionary selection of threonine unknown. Once described as belonging to the Ntn superfamily, nucleophiles in autoproteolytic systems. Conservative substitution the autoactivation of the less numerous DmpA/OAT (D/O) of the nucleophile (T282C) allowed determination of a 1.6-Å proen- superfamily has not been studied (14). -
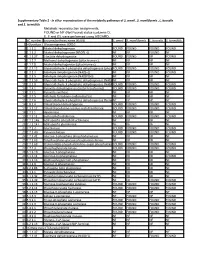
Supplementary Table 2 - in Silico Reconstruction of the Metabolic Pathways of S
Supplementary Table 2 - In silico reconstruction of the metabolic pathways of S. amnii , S. moniliformis , L. buccalis and S. termiditis Metabolic reconstruction assignments, FOUND or NF (Not Found) status (columns D, E, F and G), were performed using ASGARD, EC number Enzyme/pathway name (KEGG) S. amnii S. moniliformis L. buccalis S. termiditis 1 >Glycolysis / Gluconeogenesis 00010 2 1.1.1.1 Alcohol dehydrogenase. FOUND FOUND FOUND FOUND 3 1.1.1.2 Alcohol dehydrogenase (NADP(+)). NF NF FOUND NF 4 1.1.1.27 L-lactate dehydrogenase. FOUND FOUND NF FOUND 5 1.1.2.7 Methanol dehydrogenase (cytochrome c). NF NF NF NF 6 1.1.2.8 Alcohol dehydrogenase (cytochrome c). NF NF NF NF 7 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating).FOUND FOUND FOUND FOUND 8 1.2.1.3 Aldehyde dehydrogenase (NAD(+)). NF NF FOUND FOUND 9 1.2.1.5 Aldehyde dehydrogenase (NAD(P)(+)). NF NF NF NF 10 1.2.1.59 Glyceraldehyde-3-phosphate dehydrogenase (NAD(P)(+))NF (phosphorylating).NF NF NF 11 1.2.1.9 Glyceraldehyde-3-phosphate dehydrogenase (NADP(+)).FOUND FOUND FOUND FOUND 12 1.2.4.1 Pyruvate dehydrogenase (acetyl-transferring). FOUND FOUND FOUND FOUND 13 1.2.7.1 Pyruvate synthase. NF NF NF NF 14 1.2.7.5 Aldehyde ferredoxin oxidoreductase. NF NF NF NF 15 1.2.7.6 Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin).NF NF NF NF 16 1.8.1.4 Dihydrolipoyl dehydrogenase. FOUND FOUND FOUND FOUND 17 2.3.1.12 Dihydrolipoyllysine-residue acetyltransferase. FOUND FOUND FOUND FOUND 18 2.7.1.1 Hexokinase. -

1 Supplementary Table 1. List (Akronym and Description) Of
Supplementary Table 1. List (akronym and description) of differentially expressed genes (P < 0.05) solely related to copulation, in in pig endocervix (Cvx) up- or down-regulated. UP Description DOWN AGT angiotensinogen (serpin peptidase inhibitor, clade A, member 8) ABHD2 abhydrolase domain containing 2(ABHD2) AKAP13 A kinase (PRKA) anchor protein 13; A-kinase anchor protein 13-like AGPAT2 1-acylglycerol-3-phosphate O-acyltransferase 2(AGPAT2) AKAP9 A kinase (PRKA) anchor protein 9; A-kinase anchor protein 9-like ANXA5 annexin A5(ANXA5) ankyrin repeat domain 33B; ankyrin repeat domain-containing ANKRD33B protein 33B-like ATP6V0C ATPase H+ transporting V0 subunit c(ATP6V0C) C2H5orf45 chromosome 2 open reading frame, human C5orf45 B3GALT2 beta-1,3-galactosyltransferase 2(B3GALT2) C4H1orf110 chromosome 4 open reading frame, human C1orf110 B4GALT1 beta-1,4-galactosyltransferase 1(B4GALT1) CCDC130 coiled-coil domain containing 130(CCDC130) BRINP2 BMP/retinoic acid inducible neural specific 2(BRINP2) CCDC92 coiled-coil domain containing 92(CCDC92) C7H14orf119 chromosome 7 open reading frame, human C14orf119(C7H14orf119) CDKAL1 CDK5 regulatory subunit associated protein 1 like 1(CDKAL1) C8H4orf32 chromosome 8 open reading frame, human C4orf32 COLCA2 colorectal cancer associated 2(COLCA2) CD47 CD47 molecule CPD carboxypeptidase D(CPD) CDH7 cadherin 7(CDH7) CRTC1 CREB regulated transcription coactivator 1(CRTC1) CMPK2 cytidine/uridine monophosphate kinase 2(CMPK2) cap methyltransferase 1; cap-specific mRNA (nucleoside-2-O-)- methyltransferase 1-like; -

Female, 28 and 22 Years)
Serum proteome: Proteins identified by their tryptic peptides in samples from two healthy donors (female, 28 and 22 years) Number of proteins identified: 2669 Protein accession number protein name O14874 [3-methyl-2-oxobutanoate dehydrogenase [lipoamide]] kinase, mitochondrial precursor (EC 2.7.1.115) Q16654 [Pyruvate dehydrogenase [lipoamide]] kinase isozyme 4, mitochondrial precursor (EC 2.7.1.99) Q9P0J1 [Pyruvate dehydrogenase [Lipoamide]]-phosphatase 1, mitochondrial precursor (EC 3.1.3.43) (PDP 1) P61604 10 kDa heat shock protein, mitochondrial (Hsp10) (10 kDa chaperonin) Q15029 116 kDa U5 small nuclear ribonucleoprotein component (U5 snRNP- specific protein, 116 kDa) P62258 14-3-3 protein epsilon (14-3-3E) Q9C0C2 182 kDa tankyrase 1-binding protein Q9NRZ5 1-acyl-sn-glycerol-3-phosphate acyltransferase delta (EC 2.3.1.51) (1- AGP acyltransferase 4) Q9NRZ7 1-acyl-sn-glycerol-3-phosphate acyltransferase gamma (EC 2.3.1.51) (1- AGP acyltransferase 3) Q01970 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase beta 3 (EC 3.1.4.11) P51178 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta 1 (EC 3.1.4.11) P16885 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma 2 (EC 3.1.4.11) O75038 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase-like 4 (EC 3.1.4.11) P09543 2',3'-cyclic-nucleotide 3'-phosphodiesterase (EC 3.1.4.37) (CNP) (CNPase). Q05823 2-5A-dependent ribonuclease (EC 3.1.26.-) (2-5A-dependent RNase) (Ribonuclease L) (RNase L) Q99460 26S proteasome non-ATPase regulatory subunit 1 (26S proteasome