Browsing Genes and Genomes with Ensembl
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ABSTRACT MITCHELL III, ROBERT DRAKE. Global Human Health
ABSTRACT MITCHELL III, ROBERT DRAKE. Global Human Health Risks for Arthropod Repellents or Insecticides and Alternative Control Strategies. (Under the direction of Dr. R. Michael Roe). Protein-coding genes and environmental chemicals. New paradigms for human health risk assessment of environmental chemicals emphasize the use of molecular methods and human-derived cell lines. In this study, we examined the effects of the insect repellent DEET (N, N-diethyl-m-toluamide) and the phenylpyrazole insecticide fipronil (fluocyanobenpyrazole) on transcript levels in primary human hepatocytes. These chemicals were tested individually and as a mixture. RNA-Seq showed that 100 µM DEET significantly increased transcript levels for 108 genes and lowered transcript levels for 64 genes and fipronil at 10 µM increased the levels of 2,246 transcripts and decreased the levels for 1,428 transcripts. Fipronil was 21-times more effective than DEET in eliciting changes, even though the treatment concentration was 10-fold lower for fipronil versus DEET. The mixture of DEET and fipronil produced a more than additive effect (levels increased for 3,017 transcripts and decreased for 2,087 transcripts). The transcripts affected in our treatments influenced various biological pathways and processes important to normal cellular functions. Long non-protein coding RNAs and environmental chemicals. While the synthesis and use of new chemical compounds is at an all-time high, the study of their potential impact on human health is quickly falling behind. We chose to examine the effects of two common environmental chemicals, the insect repellent DEET and the insecticide fipronil, on transcript levels of long non-protein coding RNAs (lncRNAs) in primary human hepatocytes. -

Emerging Genetic Alterations Linked to Male Infertility: X-Chromosome Copy Number Variation and Spermatogenesis Regulatory Genes’ Expression
Central Annals of Reproductive Medicine and Treatment Bringing Excellence in Open Access Research Article *Corresponding author Susana Fernandes, Department of Genetics, Faculty of Medicine of University of Porto, Al. Prof. Hernâni Emerging Genetic Alterations Monteiro, 4200 - 319 Porto, Portugal; Tel: 351 22 551 36 47; Email: Submitted: 12 October 2016 Linked to Male Infertility: Accepted: 10 February 2017 Published: 14 February 2017 X-Chromosome Copy Number Copyright © 2017 Fernandes et al. Variation and Spermatogenesis OPEN ACCESS Keywords Regulatory Genes’ Expression • Male infertility • Spermatogenesis Catarina Costa1, Maria João Pinho1,2, Alberto Barros1,2,3, and • DNA copy number variations • Gene expression Susana Fernandes1,2* 1Department of Genetics, Faculty of Medicine of University of Porto, Portugal 2i3S – Instituto de Investigação e Inovação em Saúde, University of Porto, Portugal 3Centre for Reproductive Genetics A Barros, Portugal Abstract The etiopathogenesis of primary testicular failure remains undefined in 50% of cases. Most of these idiopathic cases probably result from genetic mutations/anomalies. Novel causes, like Copy Number Variation and gene expression profile, are being explored thanks to recent advances in the field of genetics. Our aim was to study Copy Number Variation (CNV) 67, a patient-specific CNV related to spermatogenic anomaly and evaluate the expression of regulatory genes AKAP4, responsible for sperm fibrous sheet assembly, and STAG3, essential for sister chromatid cohesion during meiosis. One hundred infertile men were tested for CNV67 with quantitative PCR (qPCR). Quantitative real-time PCR was performed to evaluate gene expression patterns of the two mentioned genes in testicular biopsies from 22 idiopathic infertile patients. CNV67 deletion was found in 2% of patients, with the same semen phenotype described in previous studies. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Reduced Transfer Coefficient of Carbon Monoxide in Pulmonary Arterial Hypertension Implicates Rare Protein-Truncating Variants I
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.11.871210; this version posted December 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Reduced transfer coefficient of carbon monoxide in pulmonary arterial hypertension implicates rare protein- truncating variants in KDR Genotype-phenotype inference reveals novel PAH risk genes Emilia M. Swietlik, M.D.[1], Daniel Greene, PhD[2,3], Na Zhu, PhD[4,5], Karyn Megy, PhD[2,3], Marcella Cogliano, M.Sc.[6], Smitha Rajaram, M.D.[7], Divya Pandya, M.Sc.[1], Tobias Tilly, M.Sc.[1], Katie A. Lutz, BS[8], Carrie C. L. Welch, PhD[4], Michael W. Pauciulo, BS, MBA[8,9], Laura Southgate, PhD[10], Jennifer M. Martin, M.St.[3], Carmen M. Treacy, B.Sc.[1], Harm J. Bogaard, M.D., PhD[11], Colin Church, PhD[12], Gerry Coghlan, M.D.[13], Anna W. Coleman, MS[8], Robin Condliffe, M.D.[14], Mélanie Eyries, PhD[15], Henning Gall, M.D., PhD[16], Stefano Ghio, M.D.[17], Simon Holden, PhD[18], Luke Howard, M.D., PhD.[19], David G. Kiely, M.D.[14], Gabor Kovacs, M.D.[20,21], Jim Lordan, PhD[22], Rajiv D. Machado, PhD[10], Robert V. MacKenzie Ross, M.B, B.Chir[23], Shahin Moledina, M.B.Ch.B.[24], David Montani, M.D., PhD[25], Horst Olschewski, M.D.[20,21], Joanna Pepke-Zaba, PhD[26], Christopher J. -

Type of the Paper (Article
Article Differential Expression Profile of lncRNAs from Primary Human Hepatocytes Following DEET and Fipronil Exposure Robert D. Mitchell III 1, Andrew D. Wallace 2, Ernest Hodgson 2,3 and R. Michael Roe 1,* 1 Department of Entomology and Plant Pathology, North Carolina Agromedicine Institute, Campus Box 7647, 3230 Ligon Street, North Carolina State University, Raleigh, NC 27695, USA; [email protected] (R.D.M.) 2 Toxicology Program, Department of Biology, North Carolina State University, Raleigh, NC 27695, USA; [email protected] (A.D.W.); [email protected] (E.H.) 3 Department of Applied Ecology, Toxicology Program, Department of Biology, North Carolina Agromedicine Institute, North Carolina State University, Raleigh, NC 27695, USA * Correspondence: [email protected]; Tel.: +1-919-515-4325 Received: 15 August 2017; Accepted: 4 October 2017; Published: 7 October 2017 Abstract: While the synthesis and use of new chemical compounds is at an all-time high, the study of their potential impact on human health is quickly falling behind, and new methods are needed to assess their impact. We chose to examine the effects of two common environmental chemicals, the insect repellent N,N-diethyl-m-toluamide (DEET) and the insecticide fluocyanobenpyrazole (fipronil), on transcript levels of long non-protein coding RNAs (lncRNAs) in primary human hepatocytes using a global RNA-Seq approach. While lncRNAs are believed to play a critical role in numerous important biological processes, many still remain uncharacterized, and their functions and modes of action remain largely unclear, especially in relation to environmental chemicals. RNA- Seq showed that 100 µM DEET significantly increased transcript levels for 2 lncRNAs and lowered transcript levels for 18 lncRNAs, while fipronil at 10 µM increased transcript levels for 76 lncRNAs and decreased levels for 193 lncRNAs. -
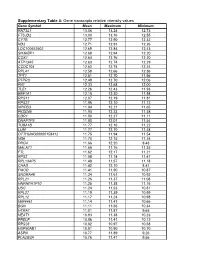
Gene Transcripts Relative Intensity Values
Supplementary Table 4: Gene transcripts relative intensity values Gene Symbol Mean Maximum Minimum RN7SL1 13.06 13.24 12.73 FTSJD2 13.00 13.16 12.55 CYTB 12.77 12.90 12.32 ND2 12.71 12.91 12.26 LOC100652902 12.69 12.84 12.43 SH3KBP1 12.68 12.94 12.20 COX1 12.64 12.76 12.20 ATP13A5 12.63 12.74 12.29 CCDC104 12.62 12.74 12.24 RPL41 12.58 12.66 12.36 TPT1 12.51 12.70 11.96 PTPRO 12.49 12.70 12.06 FN1 12.33 12.68 12.00 TLE1 12.26 12.43 11.93 EEF1A1 12.15 12.30 11.98 RPS11 12.07 12.19 11.81 RPS27 11.96 12.10 11.72 NPIPB3 11.94 12.21 11.65 FKSG49 11.94 12.23 11.58 CDR1 11.90 12.27 11.11 DNAPTP3 11.80 12.07 11.54 TUBA1B 11.77 12.16 11.22 LUM 11.77 12.10 11.48 OTTHUMG00000158412 11.75 11.94 11.54 ND6 11.70 12.15 11.24 PRG4 11.66 12.20 9.43 MALAT1 11.65 11.76 11.35 FTL 11.62 12.17 11.21 RPS2 11.58 11.74 11.47 RPL13AP5 11.48 11.57 11.18 CHAD 11.42 12.10 8.41 FMOD 11.41 11.90 10.87 SNORA48 11.34 11.61 10.92 RPL21 11.26 11.37 11.08 HNRNPA1P10 11.25 11.38 11.16 UBC 11.24 11.53 10.81 RPL27 11.19 11.39 10.99 RPL12 11.17 11.24 10.98 MIR4461 11.14 11.47 10.66 BGN 11.11 11.66 10.34 HTRA1 11.01 11.87 9.65 NEAT1 10.93 11.38 10.23 PRELP 10.86 11.41 10.12 RPS28 10.82 10.97 10.58 HSP90AB1 10.81 10.90 10.70 ASPN 10.77 11.89 9.26 PLA2G2A 10.76 11.47 9.56 H3F3A 10.69 10.86 10.41 EEF1G 10.69 10.94 10.11 C6orf48 10.64 11.02 10.35 UBA52 10.58 10.75 10.08 CTGF 10.57 11.33 9.66 MGP 10.57 11.22 10.09 YBX1 10.56 10.79 10.01 MT1X 10.56 11.89 9.88 NPC2 10.55 10.80 10.12 LAPTM4A 10.51 10.78 10.18 ITM2B 10.51 10.68 10.34 IBSP 10.50 11.50 7.17 NPIPB5 10.50 10.73 10.26 TUBA1A -

This Item Is the Archived Peer-Reviewed Author-Version Of
This item is the archived peer-reviewed author-version of: Deiodinase knockdown affects zebrafish eye development at the level of gene expression, morphology and function Reference: Houbrechts Anne M., Vergauwen Lucia, Bagci Enise, Van houcke Jolien, Heijlen Marjolein, Kulemeka Bernard, Hyde David R., Knapen Dries, Darras Veerle M..- Deiodinase knockdown affects zebrafish eye development at the level of gene expression, morphology and function Molecular and cellular endocrinology - ISSN 0303-7207 - 424(2016), p. 81-93 Full text (Publishers DOI): http://dx.doi.org/doi:10.1016/j.mce.2016.01.018 To cite this reference: http://hdl.handle.net/10067/1314550151162165141 Institutional repository IRUA 1 Deiodinase knockdown affects zebrafish eye development at the level 2 of gene expression, morphology and function. 3 Anne M. Houbrechts1*, Lucia Vergauwen2*, Enise Bagci2,3, Jolien Van houcke1, Marjolein 4 Heijlen1, Bernard Kulemeka4, David R. Hyde4, Dries Knapen2, Veerle M. Darras1 5 1 Laboratory of Comparative Endocrinology, Department of Biology, Division of 6 Animal Physiology and Neurobiology, KU Leuven, 3000 Leuven, Belgium 7 2 Zebrafishlab, Veterinary Physiology and Biochemistry, Department of Veterinary 8 Sciences, University of Antwerp, Universiteitsplein 1, 2610 Wilrijk, Belgium 9 3 Systemic Physiological & Ecotoxicological Research, Department of Biology, 10 University of Antwerp, Universiteitsplein 1, 2610 Wilrijk, Belgium 11 4 Department of Biological Sciences and Center for Zebrafish Research, University of 12 Notre Dame, Notre Dame, -

Mechanism of Ant1-Induced Human Diseases and Stress Signaling
MECHANISM OF ANT1-INDUCED HUMAN DISEASES AND STRESS SIGNALING Item Type Dissertation Authors Liu, Yaxin Rights Attribution-NonCommercial-NoDerivatives 4.0 International Download date 09/10/2021 19:50:21 Item License http://creativecommons.org/licenses/by-nc-nd/4.0/ Link to Item http://hdl.handle.net/20.500.12648/1772 MECHANISM OF ANT1-INDUCED HUMAN DISEASES AND STRESS SIGNALING Yaxin Liu A Dissertation in Department of Biochemistry and Molecular Biology Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Graduate Studies of State University of New York, Upstate Medical University. Approved Date Abstract Mechanism of Ant1-induced Human Diseases and Stress Signaling Yaxin Liu Xin Jie Chen Adenine nucleotide translocase (Ant) is a mitochondrial inner membrane protein, the primary function of which is to mediate the ADP/ATP exchange across the inner membrane. Missense mutations in Ant1, the skeletal muscle- and heart-specific isoform, induce human disorders including autosomal dominant Progressive External Opthalmoplegia (adPEO), cardiomyopathy and myopathy. Several models were proposed to interpret the pathogenesis of mutant Ant1-induced diseases, but no consensus has been reached. Our lab has previously found that mutant Aac2, the homolog of Ant1 in yeast, causes cell death due to the mitochondrial biogenesis defect. In the present study, we provided biochemical evidence supporting the idea that the mutant Aac2 proteins are misfolded, which derails the proteostasis on the inner membrane. We found that the assembly and stability of multiple protein complexes on the inner membrane are affected, including those involved in mitochondrial respiration and protein transport. -

A Brain-Derived Neurotrophic Factor
www.nature.com/scientificreports OPEN A Brain-Derived Neurotrophic Factor Mimetic Is Sufcient to Restore Cone Photoreceptor Visual Received: 17 January 2017 Accepted: 25 August 2017 Function in an Inherited Blindness Published: xx xx xxxx Model Conor Daly1, Lisa Shine1, Theresa Hefernan1, Sudhakar Deeti1, Alison L. Reynolds 1, John J. O’Connor1, Eugène T. Dillon 1,2, David J. Dufy 3,5, Walter Kolch 1,3,4, Gerard Cagney1 & Breandán N. Kennedy 1 Controversially, histone deacetylase inhibitors (HDACi) are in clinical trial for the treatment of inherited retinal degeneration. Utilizing the zebrafsh dyeucd6 model, we determined if treatment with HDACi can rescue cone photoreceptor-mediated visual function. dye exhibit defective visual behaviour and retinal morphology including ciliary marginal zone (CMZ) cell death and decreased photoreceptor outer segment (OS) length, as well as gross morphological defects including hypopigmentation and pericardial oedema. HDACi treatment of dye results in signifcantly improved optokinetic (OKR) (~43 fold, p < 0.001) and visualmotor (VMR) (~3 fold, p < 0.05) responses. HDACi treatment rescued gross morphological defects and reduced CMZ cell death by 80%. Proteomic analysis of dye eye extracts suggested BDNF-TrkB and Akt signaling as mediators of HDACi rescue in our dataset. Co- treatment with the TrkB antagonist ANA-12 blocked HDACi rescue of visual function and associated Akt phosphorylation. Notably, sole treatment with a BDNF mimetic, 7,8-dihydroxyfavone hydrate, signifcantly rescued dye visual function (~58 fold increase in OKR, p < 0.001, ~3 fold increase in VMR, p < 0.05). In summary, HDACi and a BDNF mimetic are sufcient to rescue retinal cell death and visual function in a vertebrate model of inherited blindness. -
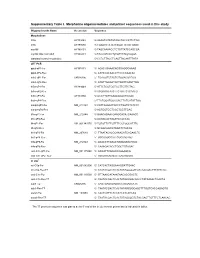
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

Copia De 2019 Tesis Drmed INDICE
UNIVERSIDAD AUTÓNOMA DE NUEVO LEÓN FACULTAD DE MEDICINA ANÁLISIS DE EXOMA EN DOS FAMILIAS NUCLEARES CON GASTROSQUISIS RECURRENTE: ESTUDIO PILOTO PARA LA IDENTIFICACIÓN DE GENES CANDIDATOS POR VÍCTOR MICHAEL SALINAS TORRES COMO REQUISITO PARA OBTENER EL GRADO DE DOCTOR EN MEDICINA JULIO, 2019 UNIVERSIDAD AUTÓNOMA DE NUEVO LEÓN FACULTAD DE MEDICINA DEPARTAMENTO DE GENÉTICA ANÁLISIS DE EXOMA EN DOS FAMILIAS NUCLEARES CON GASTROSQUISIS RECURRENTE: ESTUDIO PILOTO PARA LA IDENTIFICACIÓN DE GENES CANDIDATOS POR VÍCTOR MICHAEL SALINAS TORRES COMO REQUISITO PARA OBTENER EL GRADO DE DOCTOR EN MEDICINA JULIO, 2019 ANÁLISIS DE EXOMA EN DOS FAMILIAS NUCLEARES CON GASTROSQUISIS RECURRENTE: ESTUDIO PILOTO PARA LA IDENTIFICACIÓN DE GENES CANDIDATOS Aprobación de la tesis: ___________________________________ Dr. med. Laura Elia Martínez de Villarreal Directora de la tesis ___________________________________ Dr. med. Luis Daniel Campos Acevedo Miembro ___________________________________ Dr. med. María Esthela Morales Pérez Miembro ____________________________________ Dr. med. Erika del Carmen Ochoa Correa Miembro ____________________________________ Dr. C. Hugo Leonid Gallardo Blanco Miembro ____________________________________ Dr. med. Felipe Arturo Morales Martínez Subdirector de Estudios de Posgrado ii! FINANCIAMIENTO El presente proyecto de investigación fue financiado con recursos económicos propios del Departamento de Genética. Responsable: Dr. med. Laura Elia Martínez de Villarreal. APROBACIÓN DEL COMITÉ DE ÉTICA Y COMITÉ DOCTORAL El presente proyecto de investigación fue aprobado por el Comité de Ética y el Comité Doctoral de la Facultad de Medicina y Hospital Universitario “Dr. José Eleuterio González” de la Universidad Autónoma de Nuevo León. No. GN17-00002 (21 de septiembre de 2017). No. FMSEPG-3322/1017 (25 de octubre de 2017). iii! AGRADECIMIENTOS A Dios por permitirme tener y disfrutar a mi Familia, gracias a todos y cada uno de Ustedes por apoyarme en cada decisión y proyecto, por permitirme cumplir con excelencia en el desarrollo de esta tesis. -

Identification of Cancer-Specific Transcripts
Identification of cancer-specific transcripts: With emphasis on the hunt for fusion genes in colorectal cancer Marthe Eken Thesis for the master’s degree in Molecular Bioscience at Department of Molecular Bioscience (IMBV), Faculty of Mathematics and Natural sciences UNIVERSITY OF OSLO December 2008 2 Acknowledgements This work was carried out in the project Group of Genome Biology, at the Department of Cancer Prevention, Rikshospitalet-Radiumhospitalet Medical Center, from March 2007 to December 2008. First of all, I would like to thank my supervisor, Rolf I. Skotheim, for his great support throughout the project, for always taking the time to answer my many questions, and for his everlasting patience with my forgotten italics. I also wish to thank my co-supervisor and head of the department, Ragnhild A. Lothe, for giving me the opportunity to be part of such an excellent group. I greatly appreciate the other members of the department for making this a great place to work, especially Guro for being my lab-oracle, Anita for providing microarray data, Zere for helping me with the cloning, and Hilde for our many discussions. I am grateful to my mother for all her great advices through the years and for always being there for me, and to my father for telling me I could do anything. Finally, special thanks go to my fiancé, Joachim, for his overwhelming patience and support, for always believing in me, and for making me believe in myself. Oslo, December 2008 Marthe Eken Table of contents TABLE OF CONTENTS..................................................................................................................... 4 SUMMARY .......................................................................................................................................... 6 ABBREVIATIONS .............................................................................................................................. 8 GENE SYMBOLS ...............................................................................................................................