Identification of Breast Cancer Subtype-Specific Biomarkers By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exploring Prostate Cancer Genome Reveals Simultaneous Losses of PTEN, FAS and PAPSS2 in Patients with PSA Recurrence After Radical Prostatectomy
Int. J. Mol. Sci. 2015, 16, 3856-3869; doi:10.3390/ijms16023856 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Exploring Prostate Cancer Genome Reveals Simultaneous Losses of PTEN, FAS and PAPSS2 in Patients with PSA Recurrence after Radical Prostatectomy Chinyere Ibeawuchi 1, Hartmut Schmidt 2, Reinhard Voss 3, Ulf Titze 4, Mahmoud Abbas 5, Joerg Neumann 6, Elke Eltze 7, Agnes Marije Hoogland 8, Guido Jenster 9, Burkhard Brandt 10 and Axel Semjonow 1,* 1 Prostate Center, Department of Urology, University Hospital Muenster, Albert-Schweitzer-Campus 1, Gebaeude 1A, Muenster D-48149, Germany; E-Mail: [email protected] 2 Center for Laboratory Medicine, University Hospital Muenster, Albert-Schweitzer-Campus 1, Gebaeude 1A, Muenster D-48149, Germany; E-Mail: [email protected] 3 Interdisciplinary Center for Clinical Research, University of Muenster, Albert-Schweitzer-Campus 1, Gebaeude D3, Domagkstrasse 3, Muenster D-48149, Germany; E-Mail: [email protected] 4 Pathology, Lippe Hospital Detmold, Röntgenstrasse 18, Detmold D-32756, Germany; E-Mail: [email protected] 5 Institute of Pathology, Mathias-Spital-Rheine, Frankenburg Street 31, Rheine D-48431, Germany; E-Mail: [email protected] 6 Institute of Pathology, Klinikum Osnabrueck, Am Finkenhuegel 1, Osnabrueck D-49076, Germany; E-Mail: [email protected] 7 Institute of Pathology, Saarbrücken-Rastpfuhl, Rheinstrasse 2, Saarbrücken D-66113, Germany; E-Mail: [email protected] 8 Department -

Ten Commandments for a Good Scientist
Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Wageningen 2010 Thesis committee Thesis supervisors Prof. dr. ir. Ivonne M.C.M. Rietjens Professor of Toxicology Wageningen University Prof. dr. Albertinka J. Murk Personal chair at the sub-department of Toxicology Wageningen University Thesis co-supervisor Dr. ir. Jacques J.M. Vervoort Associate professor at the Laboratory of Biochemistry Wageningen University Other members Prof. dr. Michael R. Muller, Wageningen University Prof. dr. ir. Huub F.J. Savelkoul, Wageningen University Prof. dr. Everardus J. van Zoelen, Radboud University Nijmegen Dr. ir. Toine F.H. Bovee, RIKILT, Wageningen This research was conducted under the auspices of the Graduate School VLAG Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Thesis submitted in fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Tuesday 14 September 2010 at 4 p.m. in the Aula Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds. Ana María Sotoca Covaleda Thesis Wageningen University, Wageningen, The Netherlands, 2010, With references, and with summary in Dutch. ISBN: 978-90-8585-707-5 “Caminante no hay camino, se hace camino al andar. Al andar se hace camino, y al volver la vista atrás se ve la senda que nunca se ha de volver a pisar” - Antonio Machado – A mi madre. -

A Dissertation Entitled the Androgen Receptor
A Dissertation entitled The Androgen Receptor as a Transcriptional Co-activator: Implications in the Growth and Progression of Prostate Cancer By Mesfin Gonit Submitted to the Graduate Faculty as partial fulfillment of the requirements for the PhD Degree in Biomedical science Dr. Manohar Ratnam, Committee Chair Dr. Lirim Shemshedini, Committee Member Dr. Robert Trumbly, Committee Member Dr. Edwin Sanchez, Committee Member Dr. Beata Lecka -Czernik, Committee Member Dr. Patricia R. Komuniecki, Dean College of Graduate Studies The University of Toledo August 2011 Copyright 2011, Mesfin Gonit This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract of The Androgen Receptor as a Transcriptional Co-activator: Implications in the Growth and Progression of Prostate Cancer By Mesfin Gonit As partial fulfillment of the requirements for the PhD Degree in Biomedical science The University of Toledo August 2011 Prostate cancer depends on the androgen receptor (AR) for growth and survival even in the absence of androgen. In the classical models of gene activation by AR, ligand activated AR signals through binding to the androgen response elements (AREs) in the target gene promoter/enhancer. In the present study the role of AREs in the androgen- independent transcriptional signaling was investigated using LP50 cells, derived from parental LNCaP cells through extended passage in vitro. LP50 cells reflected the signature gene overexpression profile of advanced clinical prostate tumors. The growth of LP50 cells was profoundly dependent on nuclear localized AR but was independent of androgen. Nevertheless, in these cells AR was unable to bind to AREs in the absence of androgen. -

Delineation of Key Regulatory Elements Identifies Points Of
DELINEATION OF KEY REGULATORY ELEMENTS IDENTIFIES POINTS OF VULNERABILITY IN THE MITOGEN-ACTIVATED SIGNALING NETWORK SUPPLEMENTARY MATERIALS List of contents Supplementary Figures with legends 1. Figure S1: Distribution of primary siRNA screen data, and standardization of assay procedure. 2. Figure S2: Scatter plot of screen data. 3. Figure S3: Functional relevance of the identified targets and Calculation of residence time from PDT and cell cycle distribution. 4. Figure S4: FACS profiles for ABL1 and AKT1. Table for data in Figure 5B. 5. Figure S5: Venn diagram showing the results of the comparative analysis of other screen results 6. Figure S6: Dose response profiles for the AKT1 + ABL1 inhibitor combination for CH1, list of the 14 cell lines and their description, effect of ABL1+AKT1 inhibitor combination on increase in apoptotic cells and G1 arrest in 14 cell lines, effects of CHEK1 inhibitor on combination C1,C2 on 4 cell lines. Supplementary Tables 1. Table S1: siRNA screen results for targeted kinases and phosphatases. 2. Table S2: Gene expression status of the validated hits. 3. Table S3: Role played by identified RNAi hits in regulation of cell cycle, the effect on PDTs along with phase-specific RTs. 4. Table S4: List of molecules classified as cell cycle targets. 5. Table S5: High confidence network used for graph theory analysis. 6. Table S6: Occurrences of nodes in shortest path networks. 7. Table S7: Network file used as SNAVI background. 8. Table S8: Classification of nodes present in modules according to specificity. Legends for tables Supplementary Experimental Procedures References Figure S1 A 450 400 G1 S 350 G2 300 250 200 150 100 50 Distribution of molecules Distribution 0 -6-4-20246 Z-score 350 200 400 G1 S 300 G2 150 300 250 200 100 200 150 100 50 100 Distribution of molecules 50 0 0 0 -4 -2 0 2 4 -4-20246 -4-20246 Z-score B PLK1 GAPDH PLCg BTK PLCg CDC2A PLCg CHEK1 PLCg MET Distribution profiles of complete primary screen and western blots showing knockdown efficiency. -

Characterisation of Isomirs in Stem Cells
Characterisation of isomiRs in stem cells Geok Chin Tan Institute of Reproductive and Developmental Biology Department of Surgery and Cancer Faculty of Medicine Imperial College London Thesis submitted to Imperial College London for the degree of Doctor of Philosophy 1 Statement of Originality All experiments included in this thesis were performed by me unless otherwise stated in the text. 2 Copyright Statement ‘The copyright of this thesis rests with the author and is made available under a Creative Commons Attribution Non-Commercial No Derivatives licence. Researchers are free to copy, distribute or transmit the thesis on the condition that they attribute it, that they do not use it for commercial purposes and that they do not alter, transform or build upon it. For any reuse or redistribution, researchers must make clear to others the licence terms of this work’ 3 Acknowledgements I would like to thank my supervisor Dr Nicholas Dibb for giving me the opportunity to work in his lab and for all of his guidance and support throughout my PhD, without which this project would not have been possible. I am also very grateful to Dr Wei Cui for teaching me the technique of stem cell culture, her comments on my project related to stem cells and as a wonderful co-supervisor. I would like to also thank Professor Malcolm Parker for his supports and advise on academic and non-academic related subjects. Many thanks to Elcie Chan for the generation of all the stem cell libraries which forms the platform for my project. My sincere thanks also to Gunter Meister for supplying the Argonaute antibodies, Leandro Castellano for the help in the design of RNA sponges, Laki Buluwela for the pTRIPz lentiviral vector and last but not least Alywn Dart from Charlotte Bevan group for the prostate cancer cell lines. -
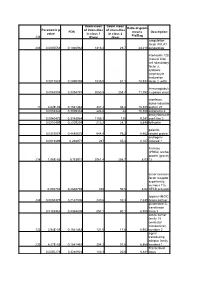
238 Parametric P- Value FDR Geom Mean of Intensities in Class 1 (Plate
Geom mean Geom mean Ratio of geom Parametric p- of intensities of intensities FDR means Description value in class 1 in class 2 Pla/Bag 238 (Plate) (Bag) coagulation factor XIII, A1 206 0,0005558 0,1940962 1313,3 29,7 44,219 polypeptide interleukin 12B (natural killer cell stimulatory factor 2, cytotoxic lymphocyte maturation 0,0017883 0,2490398 1535,5 81,1 18,933 factor 2, p40) Immunoglobuli 0,0063804 0,3394787 3050,5 258,7 11,792 n epsilon chain interferon, alpha-inducible 11 4,62E-05 0,1541482 347,2 33,4 10,395 protein 27 0,0035346 0,3039332 226,8 22 10,309 astrotactin 2 deoxyribonucle 0,0040473 0,3148984 1155,1 125 9,241 ase I-like 3 0,0010495 0,2205294 213,2 24,1 8,846 follistatin galectin- 0,0315078 0,4885675 644,8 76,2 8,462 related protein androgen- 0,0013899 0,238971 287 35,4 8,107 induced 1 A kinase (PRKA) anchor protein (gravin) 236 1,06E-05 0,115911 2061,4 256,7 8,03 12 tumor necrosis factor receptor superfamily, member 11a, 0,002746 0,2846759 405 50,5 8,02 NFKB activator lipoma HMGIC 248 0,0008375 0,2147006 240,6 32,3 7,449 fusion partner glutathione S- transferase 0,0183462 0,4368809 557,1 80,1 6,955 theta 1 solute carrier family 18 (vesicular monoamine), 122 3,94E-05 0,1541482 121,5 17,6 6,903 member 2 signal transducing adaptor family 233 6,27E-05 0,1541482 354,3 51,6 6,866 member 1 Transcribed 0,0050276 0,3240923 140,3 20,6 6,811 locus aldehyde dehydrogenas e 5 family, member A1 (succinate- semialdehyde dehydrogenas 0,0294865 0,4819829 236,6 38,2 6,194 e) guanylate cyclase activator 1A 0,0074099 0,3504479 339,1 55 6,165 -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Mrna Expression in Human Leiomyoma and Eker Rats As Measured by Microarray Analysis
Table 3S: mRNA Expression in Human Leiomyoma and Eker Rats as Measured by Microarray Analysis Human_avg Rat_avg_ PENG_ Entrez. Human_ log2_ log2_ RAPAMYCIN Gene.Symbol Gene.ID Gene Description avg_tstat Human_FDR foldChange Rat_avg_tstat Rat_FDR foldChange _DN A1BG 1 alpha-1-B glycoprotein 4.982 9.52E-05 0.68 -0.8346 0.4639 -0.38 A1CF 29974 APOBEC1 complementation factor -0.08024 0.9541 -0.02 0.9141 0.421 0.10 A2BP1 54715 ataxin 2-binding protein 1 2.811 0.01093 0.65 0.07114 0.954 -0.01 A2LD1 87769 AIG2-like domain 1 -0.3033 0.8056 -0.09 -3.365 0.005704 -0.42 A2M 2 alpha-2-macroglobulin -0.8113 0.4691 -0.03 6.02 0 1.75 A4GALT 53947 alpha 1,4-galactosyltransferase 0.4383 0.7128 0.11 6.304 0 2.30 AACS 65985 acetoacetyl-CoA synthetase 0.3595 0.7664 0.03 3.534 0.00388 0.38 AADAC 13 arylacetamide deacetylase (esterase) 0.569 0.6216 0.16 0.005588 0.9968 0.00 AADAT 51166 aminoadipate aminotransferase -0.9577 0.3876 -0.11 0.8123 0.4752 0.24 AAK1 22848 AP2 associated kinase 1 -1.261 0.2505 -0.25 0.8232 0.4689 0.12 AAMP 14 angio-associated, migratory cell protein 0.873 0.4351 0.07 1.656 0.1476 0.06 AANAT 15 arylalkylamine N-acetyltransferase -0.3998 0.7394 -0.08 0.8486 0.456 0.18 AARS 16 alanyl-tRNA synthetase 5.517 0 0.34 8.616 0 0.69 AARS2 57505 alanyl-tRNA synthetase 2, mitochondrial (putative) 1.701 0.1158 0.35 0.5011 0.6622 0.07 AARSD1 80755 alanyl-tRNA synthetase domain containing 1 4.403 9.52E-05 0.52 1.279 0.2609 0.13 AASDH 132949 aminoadipate-semialdehyde dehydrogenase -0.8921 0.4247 -0.12 -2.564 0.02993 -0.32 AASDHPPT 60496 aminoadipate-semialdehyde -

STAT3 Targets Suggest Mechanisms of Aggressive Tumorigenesis in Diffuse Large B Cell Lymphoma
STAT3 Targets Suggest Mechanisms of Aggressive Tumorigenesis in Diffuse Large B Cell Lymphoma Jennifer Hardee*,§, Zhengqing Ouyang*,1,2,3, Yuping Zhang*,4 , Anshul Kundaje*,†, Philippe Lacroute*, Michael Snyder*,5 *Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305; §Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06520; and †Department of Computer Science, Stanford University School of Engineering, Stanford, CA 94305 1The Jackson Laboratory for Genomic Medicine, Farmington, CT 06030 2Department of Biomedical Engineering, University of Connecticut, Storrs, CT 06269 3Department of Genetics and Developmental Biology, University of Connecticut Health Center, Farmington, CT 06030 4Department of Biostatistics, Yale School of Public Health, Yale University, New Haven, CT 06520 5Corresponding author: Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305. Email: [email protected] DOI: 10.1534/g3.113.007674 Figure S1 STAT3 immunoblotting and immunoprecipitation with sc-482. Western blot and IPs show a band consistent with expected size (88 kDa) of STAT3. (A) Western blot using antibody sc-482 versus nuclear lysates. Lanes contain (from left to right) lysate from K562 cells, GM12878 cells, HeLa S3 cells, and HepG2 cells. (B) IP of STAT3 using sc-482 in HeLa S3 cells. Lane 1: input nuclear lysate; lane 2: unbound material from IP with sc-482; lane 3: material IP’d with sc-482; lane 4: material IP’d using control rabbit IgG. Arrow indicates the band of interest. (C) IP of STAT3 using sc-482 in K562 cells. Lane 1: input nuclear lysate; lane 2: material IP’d using control rabbit IgG; lane 3: material IP’d with sc-482. -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

Supplemental Table 3 Two-Class Paired Significance Analysis of Microarrays Comparing Gene Expression Between Paired
Supplemental Table 3 Two‐class paired Significance Analysis of Microarrays comparing gene expression between paired pre‐ and post‐transplant kidneys biopsies (N=8). Entrez Fold q‐value Probe Set ID Gene Symbol Unigene Name Score Gene ID Difference (%) Probe sets higher expressed in post‐transplant biopsies in paired analysis (N=1871) 218870_at 55843 ARHGAP15 Rho GTPase activating protein 15 7,01 3,99 0,00 205304_s_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 6,30 4,50 0,00 1563649_at ‐‐ ‐‐ ‐‐ 6,24 3,51 0,00 1567913_at 541466 CT45‐1 cancer/testis antigen CT45‐1 5,90 4,21 0,00 203932_at 3109 HLA‐DMB major histocompatibility complex, class II, DM beta 5,83 3,20 0,00 204606_at 6366 CCL21 chemokine (C‐C motif) ligand 21 5,82 10,42 0,00 205898_at 1524 CX3CR1 chemokine (C‐X3‐C motif) receptor 1 5,74 8,50 0,00 205303_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 5,68 6,87 0,00 226841_at 219972 MPEG1 macrophage expressed gene 1 5,59 3,76 0,00 203923_s_at 1536 CYBB cytochrome b‐245, beta polypeptide (chronic granulomatous disease) 5,58 4,70 0,00 210135_s_at 6474 SHOX2 short stature homeobox 2 5,53 5,58 0,00 1562642_at ‐‐ ‐‐ ‐‐ 5,42 5,03 0,00 242605_at 1634 DCN decorin 5,23 3,92 0,00 228750_at ‐‐ ‐‐ ‐‐ 5,21 7,22 0,00 collagen, type III, alpha 1 (Ehlers‐Danlos syndrome type IV, autosomal 201852_x_at 1281 COL3A1 dominant) 5,10 8,46 0,00 3493///3 IGHA1///IGHA immunoglobulin heavy constant alpha 1///immunoglobulin heavy 217022_s_at 494 2 constant alpha 2 (A2m marker) 5,07 9,53 0,00 1 202311_s_at -

Supplementary Figure 1. Epigenomic Maps of Endocrine Therapy Sensitive and Resistant Cell Lines
Supplementary Figure 1. Epigenomic maps of endocrine therapy sensitive and resistant cell lines. (A-C) ChIP-qPCR against H3K36me3 (A), H3K4me2 (B) or FAIRE-qPCR (C) was used to validate enrichment at randomly selected sites identified by ChIP-seq specific to MCF7 (left-side panel) or LTED (right-side panel) cells. Color code represents relative fold enrichment as calculated against an internal negative control. (D) Confocal analysis of immunofluorescence assay against ERα protein in MCF7-LTED HA (>12 mo) cells. Primary (prim.) and secondary (sec.) antibody controls are shown. (E) GSC analysis of the overlap between two previously published ERα cistromes and the epigenomic maps obtained in this study. Supplementary Figure 2. ERα prognostic target genes are coherently expressed in endocrine therapy resistant cell lines. (A) RT-qPCR analysis of ERα target genes across endocrine therapy-responsive or - resistant cell lines. Expression levels are normalized against MCF7 cells (dotted line). The average and SEM of replicates are shown. (B) RT-qPCR analysis of ERα expression across endocrine therapy-responsive or -resistant cell lines. Expression levels are normalized against MCF7 cells (dotted line). The average and standard error of the mean (SEM) of replicates are shown. Supplementary Figure 3. Reprogramming of the chromatin cascade increases Notch activity in endocrine therapy resistant cell lines. (A-B) RT-qPCR analysis of two γ-secretase complex subunits across endocrine therapy- responsive or -resistant cell lines. Expression levels are normalized against MCF7 cells (dotted line). The average and SEM of replicates are shown. (C-D-E) Kaplan-Meier plots of Luminal A, Luminal B or ET patients using Notch3 expression probe.