Gene Network Analyses Unveil Possible Molecular Basis Underlying Drug‑Induced Glaucoma Ruo‑Fan Ding†, Qian Yu†, Ke Liu, Juan Du, Hua‑Jun Yin and Zhi‑Liang Ji*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
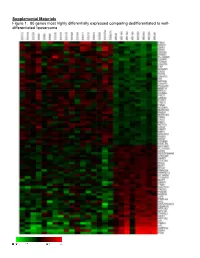
Supplemental Materials Figure 1. 80 Genes Most Highly Differentially Expressed Comparing Dedifferentiated to Well- Differentiated Liposarcoma
Supplemental Materials Figure 1. 80 genes most highly differentially expressed comparing dedifferentiated to well- differentiated liposarcoma Figure 2. Validation of microa CDC2, RACGAP1, FGFR-2, MAD2 and CITED1 200 CDK4 by Liposarcoma Subtype 16 0 12 0 80 Expression40 Level 0 Nl Fat rray data by quantitative RT-P Well Diff 16 0 p16 by Liposarcoma Subtype Dediff 12 0 80 Myxoid Expression40 Level Round Cell 0 12 0 MDM2 by Liposarcoma Subtype 10 0 Pleomorphic Nl Fat 80 60 40 Expression Level 20 RACGAP1 by LiposarcomaWell Diff Subtype 0 70 CR for genes CDK4, MDM2, p16, 60 Dediff 50 Nl Fat 40 30 Myxoid Expression20 Level Well Diff 10 50 0 Round Cell CDC2 by Liposarcoma Subtype Dediff 40 Nl Fat Pleomorphic 30 Myxoid 20 Expression Level Well Diff 10 MAD2L1 by Liposarcoma Subtype Round Cell 60 0 50 Dediff Pleomorphic 40 Nl Fat 30 Myxoid 20 Expression Level Well Diff 10 FGFR-2 by Liposarcoma Subtype 0 Round Cell 200 16 0 Dediff Nl Fat Pleomorphic 12 0 Myxoid 80 Expression Level Well Diff 40 Round Cell 0 Dediff Pleomorphic Nl Fat Myxoid Well Diff CITED1 by Liposarcoma Subtype Round Cell 10 0 80 Dediff Pleomorphic 60 40 Myxoid Expression Level 20 0 Round Cell Nl Fat Pleomorphic Well Diff Dediff Myxoid Round Cell Pleomorphic Table 1. 142 Gene classifier for liposarcoma subtype The genes used for each pair wise subtype comparison are grouped together. The flag column indicates which genes are unique to each subtype comparison. The values show the mean expression levels (actually the mean of the log expression levels was computed and than transformed back to absolute expression level). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -

G Protein-Coupled Receptors
G PROTEIN-COUPLED RECEPTORS Overview:- The completion of the Human Genome Project allowed the identification of a large family of proteins with a common motif of seven groups of 20-24 hydrophobic amino acids arranged as α-helices. Approximately 800 of these seven transmembrane (7TM) receptors have been identified of which over 300 are non-olfactory receptors (see Frederikson et al., 2003; Lagerstrom and Schioth, 2008). Subdivision on the basis of sequence homology allows the definition of rhodopsin, secretin, adhesion, glutamate and Frizzled receptor families. NC-IUPHAR recognizes Classes A, B, and C, which equate to the rhodopsin, secretin, and glutamate receptor families. The nomenclature of 7TM receptors is commonly used interchangeably with G protein-coupled receptors (GPCR), although the former nomenclature recognises signalling of 7TM receptors through pathways not involving G proteins. For example, adiponectin and membrane progestin receptors have some sequence homology to 7TM receptors but signal independently of G-proteins and appear to reside in membranes in an inverted fashion compared to conventional GPCR. Additionally, the NPR-C natriuretic peptide receptor has a single transmembrane domain structure, but appears to couple to G proteins to generate cellular responses. The 300+ non-olfactory GPCR are the targets for the majority of drugs in clinical usage (Overington et al., 2006), although only a minority of these receptors are exploited therapeutically. Signalling through GPCR is enacted by the activation of heterotrimeric GTP-binding proteins (G proteins), made up of α, β and γ subunits, where the α and βγ subunits are responsible for signalling. The α subunit (tabulated below) allows definition of one series of signalling cascades and allows grouping of GPCRs to suggest common cellular, tissue and behavioural responses. -

Multi-Functionality of Proteins Involved in GPCR and G Protein Signaling: Making Sense of Structure–Function Continuum with In
Cellular and Molecular Life Sciences (2019) 76:4461–4492 https://doi.org/10.1007/s00018-019-03276-1 Cellular andMolecular Life Sciences REVIEW Multi‑functionality of proteins involved in GPCR and G protein signaling: making sense of structure–function continuum with intrinsic disorder‑based proteoforms Alexander V. Fonin1 · April L. Darling2 · Irina M. Kuznetsova1 · Konstantin K. Turoverov1,3 · Vladimir N. Uversky2,4 Received: 5 August 2019 / Revised: 5 August 2019 / Accepted: 12 August 2019 / Published online: 19 August 2019 © Springer Nature Switzerland AG 2019 Abstract GPCR–G protein signaling system recognizes a multitude of extracellular ligands and triggers a variety of intracellular signal- ing cascades in response. In humans, this system includes more than 800 various GPCRs and a large set of heterotrimeric G proteins. Complexity of this system goes far beyond a multitude of pair-wise ligand–GPCR and GPCR–G protein interactions. In fact, one GPCR can recognize more than one extracellular signal and interact with more than one G protein. Furthermore, one ligand can activate more than one GPCR, and multiple GPCRs can couple to the same G protein. This defnes an intricate multifunctionality of this important signaling system. Here, we show that the multifunctionality of GPCR–G protein system represents an illustrative example of the protein structure–function continuum, where structures of the involved proteins represent a complex mosaic of diferently folded regions (foldons, non-foldons, unfoldons, semi-foldons, and inducible foldons). The functionality of resulting highly dynamic conformational ensembles is fne-tuned by various post-translational modifcations and alternative splicing, and such ensembles can undergo dramatic changes at interaction with their specifc partners. -

Downloaded 18 July 2014 with a 1% False Discovery Rate (FDR)
UC Berkeley UC Berkeley Electronic Theses and Dissertations Title Chemical glycoproteomics for identification and discovery of glycoprotein alterations in human cancer Permalink https://escholarship.org/uc/item/0t47b9ws Author Spiciarich, David Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California Chemical glycoproteomics for identification and discovery of glycoprotein alterations in human cancer by David Spiciarich A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Chemistry in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Carolyn R. Bertozzi, Co-Chair Professor David E. Wemmer, Co-Chair Professor Matthew B. Francis Professor Amy E. Herr Fall 2017 Chemical glycoproteomics for identification and discovery of glycoprotein alterations in human cancer © 2017 by David Spiciarich Abstract Chemical glycoproteomics for identification and discovery of glycoprotein alterations in human cancer by David Spiciarich Doctor of Philosophy in Chemistry University of California, Berkeley Professor Carolyn R. Bertozzi, Co-Chair Professor David E. Wemmer, Co-Chair Changes in glycosylation have long been appreciated to be part of the cancer phenotype; sialylated glycans are found at elevated levels on many types of cancer and have been implicated in disease progression. However, the specific glycoproteins that contribute to cell surface sialylation are not well characterized, specifically in bona fide human cancer. Metabolic and bioorthogonal labeling methods have previously enabled enrichment and identification of sialoglycoproteins from cultured cells and model organisms. The goal of this work was to develop technologies that can be used for detecting changes in glycoproteins in clinical models of human cancer. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Phototransduction Cascade Activation Steps
1 ORIGINAL RESEARCH ARTICLE Evolution of the vertebrate phototransduction cascade activation steps Trevor D. Lamb1 and David M. Hunt2,3 1Eccles Institute of Neuroscience, John Curtin School of Medical Research, The Australian National University, ACT 2600, Australia, and 2The Lions Eye Institute and 3School of Biological Sciences, The University of Western Australia, WA 6009, Australia. Correspondence: Professor Trevor Lamb <[email protected]> Keywords: Evolution; Phototransduction; Transducin; Phosphodiesterase; Cyclic nucleotide- gated channel; Opsin ABSTRACT We examine the molecular phylogeny of the proteins underlying the activation steps of vertebrate phototransduction, for both agnathan and jawed vertebrate taxa. We expand the number of taxa analysed and we update the alignment and tree building methodology from a previous analysis. For each of the four primary components (the G-protein transducin alpha subunit, GαT, the cyclic GMP phosphodiesterase, PDE6, and the alpha and beta subunits of the cGMP-gated ion channel, CNGC), the phylogenies appear consistent with expansion from an ancestral proto-vertebrate cascade during two rounds of whole-genome duplication followed by divergence of the agnathan and jawed vertebrate lineages. In each case, we consider possible scenarios for the underlying gene duplications and losses, and we apply relevant constraints to the tree construction. From tests of the topology of the resulting trees, we obtain a scenario for the expansion of each component during 2R that accurately fits the observations. Similar analysis of the visual opsins indicates that the only expansion to have occurred during 2R was the formation of Rh1 and Rh2. Finally, we propose a hypothetical scenario for the conversion of an ancestral chordate cascade into the proto-vertebrate phototransduction cascade, prior to whole-genome duplication. -

The Specificity of Downstream Signaling for A1 and A2AR
biomedicines Article The Specificity of Downstream Signaling for A1 and A2AR Does Not Depend on the C-Terminus, Despite the Importance of This Domain in Downstream Signaling Strength Abhinav R. Jain 1 , Claire McGraw 1 and Anne S. Robinson 1,2,* 1 Department of Chemical and Biomolecular Engineering, Tulane University, New Orleans, LA 70118, USA; [email protected] (A.R.J.); [email protected] (C.M.) 2 Department of Chemical Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA * Correspondence: [email protected]; Tel.: +1-412-268-7673 Received: 17 November 2020; Accepted: 9 December 2020; Published: 13 December 2020 Abstract: Recent efforts to determine the high-resolution crystal structures for the adenosine receptors (A1R and A2AR) have utilized modifications to the native receptors in order to facilitate receptor crystallization and structure determination. One common modification is a truncation of the unstructured C-terminus, which has been utilized for all the adenosine receptor crystal structures obtained to date. Ligand binding for this truncated receptor has been shown to be similar to full-length receptor for A2AR. However, the C-terminus has been identified as a location for protein-protein interactions that may be critical for the physiological function of these important drug targets. We show that variants with A2AR C-terminal truncations lacked cAMP-linked signaling compared to the full-length receptor constructs transfected into mammalian cells (HEK-293). In addition, we show that in a humanized yeast system, the absence of the full-length C-terminus affected downstream signaling using a yeast MAPK response-based fluorescence assay, though full-length receptors showed native-like G-protein coupling. -

Analysis of the Potential Correlation Between Gastric Cancer and Gastrointestinal Microbiota Via In-Silico Data Mining
Analysis of the potential correlation between gastric cancer and gastrointestinal microbiota via in-silico data mining Xuelu Ding Tianjin Medical University Yukun Zhu Tianjin Medical University Zhaoyuan She Tianjin Medical University Xuewen Liu Tianjin Medical University Cancan Zhang Tianjin First Central Hospital Xuying Ma Tianjin Medical University Feng Wang Tianjin Medical University Fei Wang Tianjin Medical University General Hospital Xin Geng ( [email protected] ) https://orcid.org/0000-0003-0604-5392 Research Keywords: bioinformatics, microbiota, gastric cancer, databases Posted Date: December 18th, 2019 DOI: https://doi.org/10.21203/rs.2.19110/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/17 Abstract Background Emerging evidence shows the gastrointestinal microbiome might play an important role in the carcinogenesis of gastric cancer. While Helicobactor pylori has been reported to be a specic risk factor of gastric cancer, it is still controversial whether signicant difference of non- H. pylori microbiota exists between gastric cancer patients and healthy control. Results In this study, we employed multiple bioinformatic databases to excavate the potential correlation between gastrointestinal microbiome and gastric cancer. The databases involved in this investigation include HMDB, STITCH, OMIM, GWAS Catalog, WebGestalt, Toppgene, GeneMANIA. In addition, the network diagrams were built by use of Cytoscape software. Notably, our results showed that 33 common genes participate in both gastrointestinal microbiome and gastric cancer. The further analysis of these common genes suggested that there was a wide array of interactions and pathways in which the correlation between gastrointestinal microbiome and gastric cancer is involved. -

GNAT3 Antibody Cat
GNAT3 Antibody Cat. No.: 55-210 GNAT3 Antibody GNAT3 antibody immunohistochemistry analysis in Flow cytometric analysis of A549 cells (right histogram) formalin fixed and paraffin embedded human colon compared to a negative control cell (left histogram).FITC- carcinoma followed by peroxidase conjugation of the conjugated goat-anti-rabbit secondary antibodies were secondary antibody and DAB staining. used for the analysis. Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human This GNAT3 antibody is generated from rabbits immunized with a KLH conjugated IMMUNOGEN: synthetic peptide between 78-105 amino acids from the Central region of human GNAT3. TESTED APPLICATIONS: Flow, IHC-P, WB For WB starting dilution is: 1:1000 APPLICATIONS: For IHC-P starting dilution is: 1:50~100 For FACS starting dilution is: 1:10~50 September 25, 2021 1 https://www.prosci-inc.com/gnat3-antibody-55-210.html PREDICTED MOLECULAR 40 kDa WEIGHT: Properties This antibody is purified through a protein A column, followed by peptide affinity PURIFICATION: purification. CLONALITY: Polyclonal ISOTYPE: Rabbit Ig CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: Supplied in PBS with 0.09% (W/V) sodium azide. CONCENTRATION: batch dependent Store at 4˚C for three months and -20˚C, stable for up to one year. As with all antibodies STORAGE CONDITIONS: care should be taken to avoid repeated freeze thaw cycles. Antibodies should not be exposed to prolonged high temperatures. Additional Info OFFICIAL SYMBOL: GNAT3 ALTERNATE NAMES: Guanine nucleotide-binding protein G(t) subunit alpha-3, Gustducin alpha-3 chain, GNAT3 ACCESSION NO.: A8MTJ3 GENE ID: 346562 USER NOTE: Optimal dilutions for each application to be determined by the researcher. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.