Bidirectional Analysis of Cryba4-Crybb1 Nascent Transcription and Nuclear Accumulation of Crybb3 Mrnas in Lens Fibers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
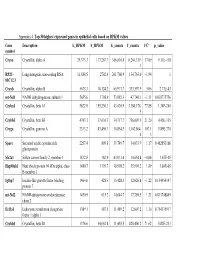
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

Supp Table 1.Pdf
Upregulated genes in Hdac8 null cranial neural crest cells fold change Gene Symbol Gene Title 134.39 Stmn4 stathmin-like 4 46.05 Lhx1 LIM homeobox protein 1 31.45 Lect2 leukocyte cell-derived chemotaxin 2 31.09 Zfp108 zinc finger protein 108 27.74 0710007G10Rik RIKEN cDNA 0710007G10 gene 26.31 1700019O17Rik RIKEN cDNA 1700019O17 gene 25.72 Cyb561 Cytochrome b-561 25.35 Tsc22d1 TSC22 domain family, member 1 25.27 4921513I08Rik RIKEN cDNA 4921513I08 gene 24.58 Ofa oncofetal antigen 24.47 B230112I24Rik RIKEN cDNA B230112I24 gene 23.86 Uty ubiquitously transcribed tetratricopeptide repeat gene, Y chromosome 22.84 D8Ertd268e DNA segment, Chr 8, ERATO Doi 268, expressed 19.78 Dag1 Dystroglycan 1 19.74 Pkn1 protein kinase N1 18.64 Cts8 cathepsin 8 18.23 1500012D20Rik RIKEN cDNA 1500012D20 gene 18.09 Slc43a2 solute carrier family 43, member 2 17.17 Pcm1 Pericentriolar material 1 17.17 Prg2 proteoglycan 2, bone marrow 17.11 LOC671579 hypothetical protein LOC671579 17.11 Slco1a5 solute carrier organic anion transporter family, member 1a5 17.02 Fbxl7 F-box and leucine-rich repeat protein 7 17.02 Kcns2 K+ voltage-gated channel, subfamily S, 2 16.93 AW493845 Expressed sequence AW493845 16.12 1600014K23Rik RIKEN cDNA 1600014K23 gene 15.71 Cst8 cystatin 8 (cystatin-related epididymal spermatogenic) 15.68 4922502D21Rik RIKEN cDNA 4922502D21 gene 15.32 2810011L19Rik RIKEN cDNA 2810011L19 gene 15.08 Btbd9 BTB (POZ) domain containing 9 14.77 Hoxa11os homeo box A11, opposite strand transcript 14.74 Obp1a odorant binding protein Ia 14.72 ORF28 open reading -

A Comprehensive Analysis of the Expression of Crystallins in Mouse Retina Jinghua Xi Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi Washington University School of Medicine in St. Louis Rafal Farjo University of Michigan - Ann Arbor Shigeo Yoshida University of Michigan - Ann Arbor Timothy S. Kern Case Western Reserve University Anand Swaroop University of Michigan - Ann Arbor See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Xi, Jinghua; Farjo, Rafal; Yoshida, Shigeo; Kern, Timothy S.; Swaroop, Anand; and Andley, Usha P., ,"A comprehensive analysis of the expression of crystallins in mouse retina." Molecular Vision.9,. 410-419. (2003). https://digitalcommons.wustl.edu/open_access_pubs/1801 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Jinghua Xi, Rafal Farjo, Shigeo Yoshida, Timothy S. Kern, Anand Swaroop, and Usha P. Andley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1801 Molecular Vision 2003; 9:410-9 <http://www.molvis.org/molvis/v9/a53> © 2003 Molecular Vision Received 28 May 2003 | Accepted 19 August 2003 | Published 28 August 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi,1 Rafal Farjo,3 Shigeo Yoshida,3 Timothy S. Kern,5 Anand Swaroop,3,4 Usha P. Andley1,2 Departments of 1Ophthalmology and Visual Sciences and 2Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. -

Related Macular Degeneration and Cutis Laxa
UvA-DARE (Digital Academic Repository) Genetic studies of age-related macular degeneration Baas, D.C. Publication date 2012 Document Version Final published version Link to publication Citation for published version (APA): Baas, D. C. (2012). Genetic studies of age-related macular degeneration. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:05 Oct 2021 G������ S������ �� A��-������� M������ D����������� D����������� M������ G������ S������ �� A��-������� | 2012 D�������� C. B��� G������ S������ �� A��-������� M������ D����������� D�������� C. B��� cover.indd 1 31-10-12 08:36 Genetic Studies of Age-related Macular Degeneration Dominique C. Baas Chapter 0.indd 1 23-10-12 19:24 The research described in this thesis was conducted at the Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences, Department of Clinical and Molecular Ophthalmogenetics, Amsterdam, The Netherlands. -

Congenital Cataracts Due to a Novel 2‑Bp Deletion in CRYBA1/A3
1614 MOLECULAR MEDICINE REPORTS 10: 1614-1618, 2014 Congenital cataracts due to a novel 2‑bp deletion in CRYBA1/A3 JING ZHANG1, YANHUA ZHANG1, FANG FANG1, WEIHONG MU1, NING ZHANG2, TONGSHUN XU3 and QINYING CAO1 1Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital; 2Department of Cardiology, The Second Hospital of Hebei Medical University; 3Department of Surgery, Shijiazhuang Obstetrics and Gynecology Hospital, Shijiazhuang, Hebei, P.R. China Received September 22, 2013; Accepted April 11, 2014 DOI: 10.3892/mmr.2014.2324 Abstract. Congenital cataracts, which are a clinically and located in the eye lens. The major human crystallins comprise genetically heterogeneous group of eye disorders, lead to 90% of protein in the mature lens and contain two different visual impairment and are a significant cause of blindness superfamilies: the small heat‑shock proteins (α-crystallins) in childhood. A major proportion of the causative mutations and the βγ-crystallins. for congenital cataracts are found in crystallin genes. In the In this study a functional candidate approach was used present study, a novel deletion mutation (c.590-591delAG) in to investigate the known crystallin genes, including CRYAA, exon 6 of CRYBA1/A3 was identified in a large family with CRYAB, CRYBA1/A3, CRYBB1, CRYBB2, CRYGC, CRYGD autosomal dominant congenital cataracts. An increase in and CRYGS, in which a major proportion of the mutations local hydrophobicity was predicted around the mutation site; identified in a large family with congenital cataracts were however, further studies are required to determine the exact found. effect of the mutation on βA1/A3-crystallin structure and function. To the best of our knowledge, this is the first report Subjects and methods of an association between a frameshift mutation in exon 6 of CRYBA1/A3 and congenital cataracts. -

Congenital Eye Disorders Gene Panel
Congenital eye disorders gene panel Contact details Introduction Regional Genetics Service Ocular conditions are highly heterogeneous and show considerable phenotypic overlap. 1 in Levels 4-6, Barclay House 2,500 children in the UK are diagnosed as blind or severely visually impaired by the time they 37 Queen Square reach one year old. As many as half of these cases are likely to be inherited and remain undiagnosed due to the vast number of genes involved in these conditions. Many congenital London, WC1N 3BH eye disorders causing visual impairment or blindness at birth or progressive visual impairment T +44 (0) 20 7762 6888 also include syndromic conditions involving additional metabolic, developmental, physical or F +44 (0) 20 7813 8578 sensory abnormalities. Gene panels offer the enhanced probability of diagnosis as a very large number of genes can be interrogated. Samples required Ocular birth defects include all inheritance modalities. Autosomal dominant and recessive 5ml venous blood in plastic EDTA diseases as well as X-linked dominant and recessive diseases are seen. These conditions can bottles (>1ml from neonates) also be caused by de novo variants. Prenatal testing must be arranged Referrals in advance, through a Clinical Genetics department if possible. Patients presenting with a phenotype appropriate for the requested sub-panel Amniotic fluid or CV samples Referrals will be accepted from clinical geneticists and consultants in ophthalmology. should be sent to Cytogenetics for Prenatal testing dissecting and culturing, with instructions to forward the sample Prenatal diagnosis may be offered as appropriate where pathogenic variants have been to the Regional Molecular Genetics identified in accordance with expected inheritance pattern and where appropriate parental laboratory for analysis testing and counselling has been conducted. -

Omics in Myopia
Journal of Clinical Medicine Review Omics in Myopia Emil Tomasz Grochowski 1,* , Karolina Pietrowska 2 , Tomasz Kowalczyk 2, Zofia Mariak 1, Adam Kretowski 2,3, Michal Ciborowski 2,* and Diana Anna Dmuchowska 1,* 1 Department of Ophthalmology, Medical University of Bialystok, M. Sklodowskiej Curie 24a, 15-276 Bialystok, Poland; [email protected] 2 Clinical Research Centre, Medical University of Bialystok, M. Sklodowskiej Curie 24a, 15-276 Bialystok, Poland; [email protected] (K.P.); [email protected] (T.K.); [email protected] (A.K.) 3 Department of Endocrinology, Diabetology and Internal Medicine, Medical University of Bialystok, M. Sklodowskiej Curie 24a, 15-276 Bialystok, Poland * Correspondence: [email protected] (E.T.G.); [email protected] (M.C.); [email protected] (D.A.D.) Received: 8 October 2020; Accepted: 25 October 2020; Published: 28 October 2020 Abstract: Myopia is a globally emerging issue, with multiple medical and socio-economic burdens and no well-established causal treatment thus far. A better insight into altered biochemical pathways and underlying pathogenesis might facilitate early diagnosis and treatment of myopia, ultimately leading to the development of more effective preventive and therapeutic measures. In this review, we summarize current data about the metabolomics and proteomics of myopia in humans and present various experimental approaches and animal models, along with their strengths and weaknesses. We also discuss the potential applicability of these -

A Novel Γd-Crystallin Mutation Causes Mild Changes in Protein Properties but Leads to Congenital Coralliform Cataract
Molecular Vision 2009; 15:1521-1529 <http://www.molvis.org/molvis/v15/a162> © 2009 Molecular Vision Received 29 April 2009 | Accepted 3 August 2009 | Published 6 August 2009 A novel γD-crystallin mutation causes mild changes in protein properties but leads to congenital coralliform cataract Li-Yun Zhang,1 Bo Gong,1 Jian-Ping Tong,2 Dorothy Shu-Ping Fan,1 Sylvia Wai-Yee Chiang,1 Dinghua Lou,2 Dennis Shun-Chiu Lam,1 Gary Hin-Fai Yam,1 Chi-Pui Pang1 1Department of Ophthalmology and Visual Sciences, The Chinese University of Hong Kong, Hong Kong, China; 2Department of Ophthalmology, the First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China Purpose: To identify the genetic lesions for congenital coralliform cataract. Methods: Two Chinese families with autosomal dominant coralliform cataract, 12 affected and 14 unaffected individuals, were recruited. Fifteen known genes associated with autosomal dominant congenital cataract were screened by two-point linkage analysis with gene based single nucleotide polymorphisms and microsatellite markers. Sequence variations were identified. Recombinant FLAG-tagged wild type or mutant γD-crystallin was expressed in human lens epithelial cells and COS-7 cells. Protein solubility and intracellular distribution were analyzed by western blotting and immunofluorescence, respectively. Results: A novel heterozygous change, c.43C>A (R15S) of γD-crystallin (CRYGD) co-segregated with coralliform cataract in one family and a known substitution, c.70C>A (P24T), in the other family. Unaffected family members and 103 unrelated control subjects did not carry these mutations. Similar to the wild type protein, R15S γD-crystallin was detergent soluble and was located in the cytoplasm. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Rescue of Retinal Degeneration by Intravitreally Injected Adult Bone Marrow–Derived Lineage-Negative Hematopoietic Stem Cells
Rescue of retinal degeneration by intravitreally injected adult bone marrow–derived lineage-negative hematopoietic stem cells Atsushi Otani, … , John Heckenlively, Martin Friedlander J Clin Invest. 2004;114(6):765-774. https://doi.org/10.1172/JCI21686. Article Stem cells Inherited retinal degenerations afflict 1 in 3,500 individuals and are a heterogeneous group of diseases that result in profound vision loss, usually the result of retinal neuronal apoptosis. Atrophic changes in the retinal vasculature are also observed in many of these degenerations. While it is thought that this atrophy is secondary to diminished metabolic demand in the face of retinal degeneration, the precise relationship between the retinal neuronal and vascular degeneration is not clear. In this study we demonstrate that whenever a fraction of mouse or human adult bone marrow– derived stem cells (lineage-negative hematopoietic stem cells [Lin– HSCs]) containing endothelial precursors stabilizes and rescues retinal blood vessels that would ordinarily completely degenerate, a dramatic neurotrophic rescue effect is also observed. Retinal nuclear layers are preserved in 2 mouse models of retinal degeneration, rd1 and rd10, and detectable, albeit severely abnormal, electroretinogram recordings are observed in rescued mice at times when they are never observed in control-treated or untreated eyes. The normal mouse retina consists predominantly of rods, but the rescued cells after treatment with Lin– HSCs are nearly all cones. Microarray analysis of rescued retinas demonstrates -

82314791.Pdf
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Am. J. Hum. Genet. 71:1216–1221, 2002 Report A Nonsense Mutation in CRYBB1 Associated with Autosomal Dominant Cataract Linked to Human Chromosome 22q Donna S. Mackay,1 Olivera B. Boskovska,1 Harry L. S. Knopf,1 Kirsten J. Lampi,3 and Alan Shiels1,2 1Departments of Ophthalmology and Visual Sciences and 2Genetics, Washington University School of Medicine, St. Louis; and 3Department of Oral Molecular Biology, Oregon Health and Science University, Portland Autosomal dominant cataract is a clinically and genetically heterogeneous lens disorder that usually presents as a sight-threatening trait in childhood. Here we have mapped dominant pulverulent cataract to the b-crystallin gene cluster on chromosome 22q11.2. Suggestive evidence of linkage was detected at markers D22S1167 (LOD score [Z] 2.09 at recombination fraction [v] 0) and D22S1154 (Z p 1.39 atv p 0 ), which closely flank the genes for bB1-crystallin (CRYBB1) and bA4-crystallin (CRYBA4). Sequencing failed to detect any nucleotide changes in CRYBA4; however, a GrT transversion in exon 6 of CRYBB1 was found to cosegregate with cataract in the family. This single-nucleotide change was predicted to introduce a translation stop codon at glycine 220 (G220X). Expression of recombinant human bB1-crystallin in bacteria showed that the truncated G220X mutant was significantly less soluble than wild type. This study has identified the first CRYBB1 mutation associated with autosomal dominant cataract in humans. Crystallin genes encode 195% of the water-soluble struc- number of evolutionarily diverse proteins—including tural proteins present in the vertebrate crystalline lens, bacterial spore-coat protein S, slime mold spherulin 3a, accounting for 130% of its mass (for review, see Graw and amphibian epidermis differentiation-specific protein— 1997). -

Novel Mutation in the Γ-S Crystallin Gene Causing Autosomal Dominant Cataract
Molecular Vision 2009; 15:476-481 <http://www.molvis.org/molvis/v15/a48> © 2009 Molecular Vision Received 24 December 2008 | Accepted 27 February 2009 | Published 4 March 2009 Novel mutation in the γ-S crystallin gene causing autosomal dominant cataract Vanita Vanita,1 Jai Rup Singh,1 Daljit Singh,2 Raymonda Varon,3 Karl Sperling3 1Centre for Genetic Disorders, Guru Nanak Dev University, Amritsar, India; 2Dr. Daljit Singh Eye Hospital, Amritsar, India; 3Institute of Human Genetics, Charitè, University Medicine of Berlin, Berlin, Germany Purpose: To identify the underlying genetic defect in a north Indian family with seven members in three-generations affected with bilateral congenital cataract. Methods: Detailed family history and clinical data were recorded. Linkage analysis using fluorescently labeled microsatellite markers for the already known candidate gene loci was performed in combination with mutation screening by bidirectional sequencing. Results: Affected individuals had bilateral congenital cataract. Cataract was of opalescent type with the central nuclear region denser than the periphery. Linkage was excluded for the known cataract candidate gene loci at 1p34–36, 1q21–25 (gap junction protein, alpha 8 [GJA8]), 2q33–36 (crystallin, gamma A [CRYGA], crystallin, gamma B [CRYGB], crystallin, gamma C [CRYGC], crystallin, gamma D [CRYGD], crystallin, beta A2 [CRYBA2]), 3q21–22 (beaded filament structural protein 2, phakinin [BFSP2]), 12q12–14 (aquaporin 0 [AQP0]), 13q11–13 (gap junction protein, alpha 3 [GJA3]), 15q21– 22, 16q22–23 (v-maf musculoaponeurotic fibrosarcoma oncogene homolog [MAF], heat shock transcription factor 4 [HSF4]), 17q11–12 (crystallin, beta A1 [CRYBA1]), 17q24, 21q22.3 (crystallin, alpha A [CRYAA]), and 22q11.2 (crystallin, beta B1 [CRYBB1], crystallin, beta B2 [CRYBB2], crystallin, beta B3 [CRYBB3], crystallin, beta A4 [CRYBA4]).