(12) Patent Application Publication (10) Pub. No.: US 2003/0198970 A1 Roberts (43) Pub
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

CRISPR Screening of Porcine Sgrna Library Identifies Host Factors
ARTICLE https://doi.org/10.1038/s41467-020-18936-1 OPEN CRISPR screening of porcine sgRNA library identifies host factors associated with Japanese encephalitis virus replication Changzhi Zhao1,5, Hailong Liu1,5, Tianhe Xiao1,5, Zichang Wang1, Xiongwei Nie1, Xinyun Li1,2, Ping Qian2,3, Liuxing Qin3, Xiaosong Han1, Jinfu Zhang1, Jinxue Ruan1, Mengjin Zhu1,2, Yi-Liang Miao 1,2, Bo Zuo1,2, ✉ ✉ Kui Yang4, Shengsong Xie 1,2 & Shuhong Zhao 1,2 1234567890():,; Japanese encephalitis virus (JEV) is a mosquito-borne zoonotic flavivirus that causes ence- phalitis and reproductive disorders in mammalian species. However, the host factors critical for its entry, replication, and assembly are poorly understood. Here, we design a porcine genome-scale CRISPR/Cas9 knockout (PigGeCKO) library containing 85,674 single guide RNAs targeting 17,743 protein-coding genes, 11,053 long ncRNAs, and 551 microRNAs. Subsequently, we use the PigGeCKO library to identify key host factors facilitating JEV infection in porcine cells. Several previously unreported genes required for JEV infection are highly enriched post-JEV selection. We conduct follow-up studies to verify the dependency of JEV on these genes, and identify functional contributions for six of the many candidate JEV- related host genes, including EMC3 and CALR. Additionally, we identify that four genes associated with heparan sulfate proteoglycans (HSPGs) metabolism, specifically those responsible for HSPGs sulfurylation, facilitate JEV entry into porcine cells. Thus, beyond our development of the largest CRISPR-based functional genomic screening platform for pig research to date, this study identifies multiple potentially vulnerable targets for the devel- opment of medical and breeding technologies to treat and prevent diseases caused by JEV. -

DBT Gene Dihydrolipoamide Branched Chain Transacylase E2
DBT gene dihydrolipoamide branched chain transacylase E2 Normal Function The DBT gene provides instructions for making part of a group of enzymes called the branched-chain alpha-keto acid dehydrogenase (BCKD) enzyme complex. Specifically, the protein produced from the DBT gene forms a critical piece of the enzyme complex called the E2 component. The BCKD enzyme complex is responsible for one step in the normal breakdown of three protein building blocks (amino acids). These amino acids—leucine, isoleucine, and valine—are obtained from the diet. They are present in many kinds of food, particularly protein-rich foods such as milk, meat, and eggs. The BCKD enzyme complex is active in mitochondria, which are specialized structures inside cells that serve as energy-producing centers. The breakdown of leucine, isoleucine, and valine produces molecules that can be used for energy. Health Conditions Related to Genetic Changes Maple syrup urine disease More than 70 mutations in the DBT gene have been identified in people with maple syrup urine disease, most often in individuals with mild variants of the disorder. These variant forms become apparent later in infancy or childhood, and they lead to delayed development and other health problems if not treated. Mutations in the DBT gene include changes in single DNA building blocks (base pairs) and insertions or deletions of a small amount of DNA in the DBT gene. These changes disrupt the normal function of the E2 component, preventing the BCKD enzyme complex from effectively breaking down leucine, isoleucine, and valine. As a result, these amino acids and their byproducts build up in the body. -

Mouse Germ Line Mutations Due to Retrotransposon Insertions Liane Gagnier1, Victoria P
Gagnier et al. Mobile DNA (2019) 10:15 https://doi.org/10.1186/s13100-019-0157-4 REVIEW Open Access Mouse germ line mutations due to retrotransposon insertions Liane Gagnier1, Victoria P. Belancio2 and Dixie L. Mager1* Abstract Transposable element (TE) insertions are responsible for a significant fraction of spontaneous germ line mutations reported in inbred mouse strains. This major contribution of TEs to the mutational landscape in mouse contrasts with the situation in human, where their relative contribution as germ line insertional mutagens is much lower. In this focussed review, we provide comprehensive lists of TE-induced mouse mutations, discuss the different TE types involved in these insertional mutations and elaborate on particularly interesting cases. We also discuss differences and similarities between the mutational role of TEs in mice and humans. Keywords: Endogenous retroviruses, Long terminal repeats, Long interspersed elements, Short interspersed elements, Germ line mutation, Inbred mice, Insertional mutagenesis, Transcriptional interference Background promoter and polyadenylation motifs and often a splice The mouse and human genomes harbor similar types of donor site [10, 11]. Sequences of full-length ERVs can TEs that have been discussed in many reviews, to which encode gag, pol and sometimes env, although groups of we refer the reader for more in depth and general infor- LTR retrotransposons with little or no retroviral hom- mation [1–9]. In general, both human and mouse con- ology also exist [6–9]. While not the subject of this re- tain ancient families of DNA transposons, none view, ERV LTRs can often act as cellular enhancers or currently active, which comprise 1–3% of these genomes promoters, creating chimeric transcripts with genes, and as well as many families or groups of retrotransposons, have been implicated in other regulatory functions [11– which have caused all the TE insertional mutations in 13]. -

Thiamine Dependency and Related Gene Mutations: Recent Aspects
Int J Anal Bio-Sci Vol. 3, No 4 (2015) 〈Review Article〉 Thiamine dependency and related gene mutations: recent aspects Sachiko Kiuchi1, Hiroshi Ihara1, Yoshikazu Nishiguchi2, Nobue Ito3, Hiromitsu Yokota3 and Naotaka Hashizume4 Summary Thiamine dependency is an inherited metabolic disorder from which patients exhibit severe symptoms of deficiency, although they are fed more than the normal requirement of this vitamin. Deficiency symptoms can be treated with pharmacologic doses of thiamine as high as 100 to 1,000 times the Dietary Reference Values. Thiamine dependency is nowadays classified as a disorder caused by genetic mutations affecting thiamine-dependent enzymes or thiamine transporter, which transports thiamine to cells. The former mutation on enzyme is known as a pyruvate dehydrogenase complex deficiency (i.e., congenital lactic acidemia and Leigh syndrome) and a deficiency in branched- chain α-ketoacid dehydrogenase complex (i.e., maple syrup urine disease). The latter are known as mutations in thiamine transporter gene that makes protein transporting thiamine into cells (encoded by SLC19A2 and SLC19A3 genes) and protein transporting thiamine diphosphate into the mitochon- dria (encoded by SLC25A19 gene). Thiamine-responsive megaloblastic anemia (TRMA), an autosomal recessive disease, is caused by loss of functional mutation in the SLC19A2 (ThTr-1). Key words: Vitamin B1 dependency, Thiamine deficiency, Maple syrup urine disease, Megaloblastic anemia, Thiamine transporter 1. Introduction the normal daily requirement from diets, dietary supplements, or intravenous dosage. The deficiency To maintain normal carbohydrate metabolism in symptoms were better treated by taking amounts of the body, we have to ingest thiamine from our diet. thiamine 100-fold greater or more than that of the The daily required amounts are 1.4 mg (4.2 µmol) for daily requirement, but the symptoms would reappear men and 1.1 mg (3.3 µmol) for women for Japanese if the treatment was stopped4. -
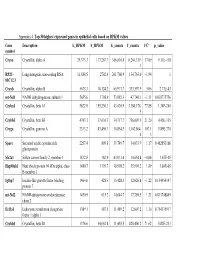
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

1 Metabolic Dysfunction Is Restricted to the Sciatic Nerve in Experimental
Page 1 of 255 Diabetes Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy Oliver J. Freeman1,2, Richard D. Unwin2,3, Andrew W. Dowsey2,3, Paul Begley2,3, Sumia Ali1, Katherine A. Hollywood2,3, Nitin Rustogi2,3, Rasmus S. Petersen1, Warwick B. Dunn2,3†, Garth J.S. Cooper2,3,4,5* & Natalie J. Gardiner1* 1 Faculty of Life Sciences, University of Manchester, UK 2 Centre for Advanced Discovery and Experimental Therapeutics (CADET), Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Sciences Centre, Manchester, UK 3 Centre for Endocrinology and Diabetes, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, UK 4 School of Biological Sciences, University of Auckland, New Zealand 5 Department of Pharmacology, Medical Sciences Division, University of Oxford, UK † Present address: School of Biosciences, University of Birmingham, UK *Joint corresponding authors: Natalie J. Gardiner and Garth J.S. Cooper Email: [email protected]; [email protected] Address: University of Manchester, AV Hill Building, Oxford Road, Manchester, M13 9PT, United Kingdom Telephone: +44 161 275 5768; +44 161 701 0240 Word count: 4,490 Number of tables: 1, Number of figures: 6 Running title: Metabolic dysfunction in diabetic neuropathy 1 Diabetes Publish Ahead of Print, published online October 15, 2015 Diabetes Page 2 of 255 Abstract High glucose levels in the peripheral nervous system (PNS) have been implicated in the pathogenesis of diabetic neuropathy (DN). However our understanding of the molecular mechanisms which cause the marked distal pathology is incomplete. Here we performed a comprehensive, system-wide analysis of the PNS of a rodent model of DN. -

Supp Table 1.Pdf
Upregulated genes in Hdac8 null cranial neural crest cells fold change Gene Symbol Gene Title 134.39 Stmn4 stathmin-like 4 46.05 Lhx1 LIM homeobox protein 1 31.45 Lect2 leukocyte cell-derived chemotaxin 2 31.09 Zfp108 zinc finger protein 108 27.74 0710007G10Rik RIKEN cDNA 0710007G10 gene 26.31 1700019O17Rik RIKEN cDNA 1700019O17 gene 25.72 Cyb561 Cytochrome b-561 25.35 Tsc22d1 TSC22 domain family, member 1 25.27 4921513I08Rik RIKEN cDNA 4921513I08 gene 24.58 Ofa oncofetal antigen 24.47 B230112I24Rik RIKEN cDNA B230112I24 gene 23.86 Uty ubiquitously transcribed tetratricopeptide repeat gene, Y chromosome 22.84 D8Ertd268e DNA segment, Chr 8, ERATO Doi 268, expressed 19.78 Dag1 Dystroglycan 1 19.74 Pkn1 protein kinase N1 18.64 Cts8 cathepsin 8 18.23 1500012D20Rik RIKEN cDNA 1500012D20 gene 18.09 Slc43a2 solute carrier family 43, member 2 17.17 Pcm1 Pericentriolar material 1 17.17 Prg2 proteoglycan 2, bone marrow 17.11 LOC671579 hypothetical protein LOC671579 17.11 Slco1a5 solute carrier organic anion transporter family, member 1a5 17.02 Fbxl7 F-box and leucine-rich repeat protein 7 17.02 Kcns2 K+ voltage-gated channel, subfamily S, 2 16.93 AW493845 Expressed sequence AW493845 16.12 1600014K23Rik RIKEN cDNA 1600014K23 gene 15.71 Cst8 cystatin 8 (cystatin-related epididymal spermatogenic) 15.68 4922502D21Rik RIKEN cDNA 4922502D21 gene 15.32 2810011L19Rik RIKEN cDNA 2810011L19 gene 15.08 Btbd9 BTB (POZ) domain containing 9 14.77 Hoxa11os homeo box A11, opposite strand transcript 14.74 Obp1a odorant binding protein Ia 14.72 ORF28 open reading -

Promiscuity and Specificity of Eukaryotic Glycosyltransferases
Biochemical Society Transactions (2020) 48 891–900 https://doi.org/10.1042/BST20190651 Review Article Promiscuity and specificity of eukaryotic glycosyltransferases Ansuman Biswas and Mukund Thattai Simons Centre for the Study of Living Machines, National Centre for Biological Sciences, TIFR, Bangalore, India Correspondence: Mukund Thattai ([email protected]) Glycosyltransferases are a large family of enzymes responsible for covalently linking sugar monosaccharides to a variety of organic substrates. These enzymes drive the synthesis of complex oligosaccharides known as glycans, which play key roles in inter-cellular interac- tions across all the kingdoms of life; they also catalyze sugar attachment during the syn- thesis of small-molecule metabolites such as plant flavonoids. A given glycosyltransferase enzyme is typically responsible for attaching a specific donor monosaccharide, via a spe- cific glycosidic linkage, to a specific moiety on the acceptor substrate. However these enzymes are often promiscuous, able catalyze linkages between a variety of donors and acceptors. In this review we discuss distinct classes of glycosyltransferase promiscuity, each illustrated by enzymatic examples from small-molecule or glycan synthesis. We high- light the physical causes of promiscuity, and its biochemical consequences. Structural studies of glycosyltransferases involved in glycan synthesis show that they make specific contacts with ‘recognition motifs’ that are much smaller than the full oligosaccharide sub- strate. There is a wide range in the sizes of glycosyltransferase recognition motifs: highly promiscuous enzymes recognize monosaccharide or disaccharide motifs across multiple oligosaccharides, while highly specific enzymes recognize large, complex motifs found on few oligosaccharides. In eukaryotes, the localization of glycosyltransferases within compartments of the Golgi apparatus may play a role in mitigating the glycan variability caused by enzyme promiscuity. -

A Comprehensive Analysis of the Expression of Crystallins in Mouse Retina Jinghua Xi Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi Washington University School of Medicine in St. Louis Rafal Farjo University of Michigan - Ann Arbor Shigeo Yoshida University of Michigan - Ann Arbor Timothy S. Kern Case Western Reserve University Anand Swaroop University of Michigan - Ann Arbor See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Xi, Jinghua; Farjo, Rafal; Yoshida, Shigeo; Kern, Timothy S.; Swaroop, Anand; and Andley, Usha P., ,"A comprehensive analysis of the expression of crystallins in mouse retina." Molecular Vision.9,. 410-419. (2003). https://digitalcommons.wustl.edu/open_access_pubs/1801 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Jinghua Xi, Rafal Farjo, Shigeo Yoshida, Timothy S. Kern, Anand Swaroop, and Usha P. Andley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1801 Molecular Vision 2003; 9:410-9 <http://www.molvis.org/molvis/v9/a53> © 2003 Molecular Vision Received 28 May 2003 | Accepted 19 August 2003 | Published 28 August 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi,1 Rafal Farjo,3 Shigeo Yoshida,3 Timothy S. Kern,5 Anand Swaroop,3,4 Usha P. Andley1,2 Departments of 1Ophthalmology and Visual Sciences and 2Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. -

Cochlin in the Eye: Functional Implications
Cochlin in the eye: Functional implications Picciani, R., Desaia, K., Guduric-Fuchs, J., Cogliati, T., Morton, C. C., & Bhattacharya, S. K. (2007). Cochlin in the eye: Functional implications. Progress in Retinal and Eye Research, 26 (5)(5), 453-469. https://doi.org/10.1016/j.preteyeres.2007.06.002 Published in: Progress in Retinal and Eye Research Queen's University Belfast - Research Portal: Link to publication record in Queen's University Belfast Research Portal General rights Copyright for the publications made accessible via the Queen's University Belfast Research Portal is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The Research Portal is Queen's institutional repository that provides access to Queen's research output. Every effort has been made to ensure that content in the Research Portal does not infringe any person's rights, or applicable UK laws. If you discover content in the Research Portal that you believe breaches copyright or violates any law, please contact [email protected]. Download date:26. Sep. 2021 Author’s Accepted Manuscript Cochlin in the eye: Functional implications Renata Picciani, Kavita Desai, Jasenka Guduric- Fuchs,Tiziana Cogliati, Cynthia C. Morton, Sanjoy K. Bhattacharya PII: S1350-9462(07)00040-7 DOI: doi:10.1016/j.preteyeres.2007.06.002 Reference: JPRR 345 www.elsevier.com/locate/prer To appear in: Progress in Retinal and Eye Research Cite this article as: Renata Picciani, Kavita Desai, Jasenka Guduric-Fuchs, Tiziana Cogliati, Cynthia C. -

Related Macular Degeneration and Cutis Laxa
UvA-DARE (Digital Academic Repository) Genetic studies of age-related macular degeneration Baas, D.C. Publication date 2012 Document Version Final published version Link to publication Citation for published version (APA): Baas, D. C. (2012). Genetic studies of age-related macular degeneration. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:05 Oct 2021 G������ S������ �� A��-������� M������ D����������� D����������� M������ G������ S������ �� A��-������� | 2012 D�������� C. B��� G������ S������ �� A��-������� M������ D����������� D�������� C. B��� cover.indd 1 31-10-12 08:36 Genetic Studies of Age-related Macular Degeneration Dominique C. Baas Chapter 0.indd 1 23-10-12 19:24 The research described in this thesis was conducted at the Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences, Department of Clinical and Molecular Ophthalmogenetics, Amsterdam, The Netherlands. -

Identification of Arylsulfatase E Mutations, Functional Analysis of Novel Missense Alleles, and Determination of Potential Phenocopies
ORIGINAL RESEARCH ARTICLE © American College of Medical Genetics and Genomics A prospective study of brachytelephalangic chondrodysplasia punctata: identification of arylsulfatase E mutations, functional analysis of novel missense alleles, and determination of potential phenocopies Claudia Matos-Miranda, MSc, MD1, Graeme Nimmo, MSc1, Bradley Williams, MGC, CGC2, Carolyn Tysoe, PhD3, Martina Owens, BS3, Sherri Bale, PhD2 and Nancy Braverman, MS, MD1,4 Purpose: The only known genetic cause of brachytelephalangic Results: In this study, 58% of males had ARSE mutations. All mutant chondrodysplasia punctata is X-linked chondrodysplasia punctata 1 alleles had negligible arylsulfatase E activity. There were no obvi- (CDPX1), which results from a deficiency of arylsulfatase E (ARSE). ous genotype–phenotype correlations. Maternal etiologies were not Historically, ARSE mutations have been identified in only 50% of reported in most patients. male patients, and it was proposed that the remainder might rep- resent phenocopies due to maternal–fetal vitamin K deficiency and Conclusion: CDPX1 is caused by loss of arylsulfatase E activ- maternal autoimmune diseases. ity. Around 40% of male patients with brachytelephalangic chon- drodysplasia punctata do not have detectable ARSE mutations or Methods: To further evaluate causes of brachytelephalangic chon- known maternal etiological factors. Improved understanding of drodysplasia punctata, we established a Collaboration Education and arylsulfatase E function is predicted to illuminate other etiologies for Test Translation program for CDPX1 from 2008 to 2010. Of the 29 brachytelephalangic chondrodysplasia punctata. male probands identified, 17 had ARSE mutations that included 10 novel missense alleles and one single-codon deletion. To determine Genet Med 2013:15(8):650–657 pathogenicity of these and additional missense alleles, we transiently expressed them in COS cells and measured arylsulfatase E activity Key Words: arylsulfatase E; brachytelephalangic chondrodysplasia using the artificial substrate, 4-methylumbelliferyl sulfate.