Congenital Eye Disorders Gene Panel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -
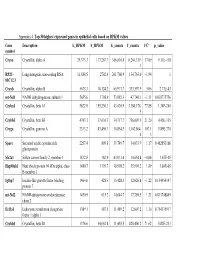
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

CRYGB Rabbit Pab
Leader in Biomolecular Solutions for Life Science CRYGB Rabbit pAb Catalog No.: A14569 Basic Information Background Catalog No. Crystallins are separated into two classes: taxon-specific, or enzyme, and ubiquitous. A14569 The latter class constitutes the major proteins of vertebrate eye lens and maintains the transparency and refractive index of the lens. Since lens central fiber cells lose their Observed MW nuclei during development, these crystallins are made and then retained throughout 21kDa life, making them extremely stable proteins. Mammalian lens crystallins are divided into alpha, beta, and gamma families; beta and gamma crystallins are also considered as a Calculated MW superfamily. Alpha and beta families are further divided into acidic and basic groups. 20kDa Seven protein regions exist in crystallins: four homologous motifs, a connecting peptide, and N- and C-terminal extensions. Gamma-crystallins are a homogeneous group of Category highly symmetrical, monomeric proteins typically lacking connecting peptides and terminal extensions. They are differentially regulated after early development. Four Primary antibody gamma-crystallin genes (gamma-A through gamma-D) and three pseudogenes (gamma-E, gamma-F, gamma-G) are tandemly organized in a genomic segment as a Applications gene cluster. Whether due to aging or mutations in specific genes, gamma-crystallins WB have been involved in cataract formation. Cross-Reactivity Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 1419 P07316 Immunogen Recombinant fusion protein containing a sequence corresponding to amino acids 80-135 of human CRYGB (NP_005201.2). Synonyms CRYGB;CRYG2;CTRCT39 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. -

Investigating Cone Photoreceptor Development Using Patient-Derived NRL Null Retinal Organoids
ARTICLE https://doi.org/10.1038/s42003-020-0808-5 OPEN Investigating cone photoreceptor development using patient-derived NRL null retinal organoids Alyssa Kallman1,11, Elizabeth E. Capowski 2,11, Jie Wang 3, Aniruddha M. Kaushik4, Alex D. Jansen2, Kimberly L. Edwards2, Liben Chen4, Cynthia A. Berlinicke3, M. Joseph Phillips2,5, Eric A. Pierce6, Jiang Qian3, ✉ ✉ Tza-Huei Wang4,7, David M. Gamm2,5,8 & Donald J. Zack 1,3,9,10 1234567890():,; Photoreceptor loss is a leading cause of blindness, but mechanisms underlying photoreceptor degeneration are not well understood. Treatment strategies would benefit from improved understanding of gene-expression patterns directing photoreceptor development, as many genes are implicated in both development and degeneration. Neural retina leucine zipper (NRL) is critical for rod photoreceptor genesis and degeneration, with NRL mutations known to cause enhanced S-cone syndrome and retinitis pigmentosa. While murine Nrl loss has been characterized, studies of human NRL can identify important insights for human retinal development and disease. We utilized iPSC organoid models of retinal development to molecularly define developmental alterations in a human model of NRL loss. Consistent with the function of NRL in rod fate specification, human retinal organoids lacking NRL develop S- opsin dominant photoreceptor populations. We report generation of two distinct S-opsin expressing populations in NRL null retinal organoids and identify MEF2C as a candidate regulator of cone development. 1 Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, USA. 2 Waisman Center, University of Wisconsin-Madison, Madison, USA. 3 Department of Ophthalmology, Wilmer Eye Institute, Johns Hopkins University School of Medicine, Baltimore, USA. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

A Comprehensive Analysis of the Expression of Crystallins in Mouse Retina Jinghua Xi Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi Washington University School of Medicine in St. Louis Rafal Farjo University of Michigan - Ann Arbor Shigeo Yoshida University of Michigan - Ann Arbor Timothy S. Kern Case Western Reserve University Anand Swaroop University of Michigan - Ann Arbor See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Xi, Jinghua; Farjo, Rafal; Yoshida, Shigeo; Kern, Timothy S.; Swaroop, Anand; and Andley, Usha P., ,"A comprehensive analysis of the expression of crystallins in mouse retina." Molecular Vision.9,. 410-419. (2003). https://digitalcommons.wustl.edu/open_access_pubs/1801 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Jinghua Xi, Rafal Farjo, Shigeo Yoshida, Timothy S. Kern, Anand Swaroop, and Usha P. Andley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1801 Molecular Vision 2003; 9:410-9 <http://www.molvis.org/molvis/v9/a53> © 2003 Molecular Vision Received 28 May 2003 | Accepted 19 August 2003 | Published 28 August 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi,1 Rafal Farjo,3 Shigeo Yoshida,3 Timothy S. Kern,5 Anand Swaroop,3,4 Usha P. Andley1,2 Departments of 1Ophthalmology and Visual Sciences and 2Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. -

Related Macular Degeneration and Cutis Laxa
UvA-DARE (Digital Academic Repository) Genetic studies of age-related macular degeneration Baas, D.C. Publication date 2012 Document Version Final published version Link to publication Citation for published version (APA): Baas, D. C. (2012). Genetic studies of age-related macular degeneration. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:05 Oct 2021 G������ S������ �� A��-������� M������ D����������� D����������� M������ G������ S������ �� A��-������� | 2012 D�������� C. B��� G������ S������ �� A��-������� M������ D����������� D�������� C. B��� cover.indd 1 31-10-12 08:36 Genetic Studies of Age-related Macular Degeneration Dominique C. Baas Chapter 0.indd 1 23-10-12 19:24 The research described in this thesis was conducted at the Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences, Department of Clinical and Molecular Ophthalmogenetics, Amsterdam, The Netherlands. -

Method for Diagnosing Non-Small Cell Lung Cancers
(19) TZZ Z _ T (11) EP 2 270 221 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 05.01.2011 Bulletin 2011/01 C12Q 1/68 (2006.01) C12N 15/12 (2006.01) G01N 33/50 (2006.01) C12N 15/11 (2006.01) (2006.01) (2006.01) (21) Application number: 10010329.0 A61P 35/00 A61K 39/00 (22) Date of filing: 22.09.2003 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Nakamura, Yusuke HU IE IT LI LU MC NL PT RO SE SI SK TR Yokohama-shi Kanagawa 225-0011 (JP) (30) Priority: 30.09.2002 US 414673 P • Daigo, Yataro 28.02.2003 US 451374 P Yokohama-shi 28.04.2003 US 466100 P Kanagawa 222-0031 (JP) • Nakatsuru, Shuichi (62) Document number(s) of the earlier application(s) in Saitama-shi accordance with Art. 76 EPC: Saitama 338-0002 (JP) 06022167.8 / 1 743 947 03753941.8 / 1 551 998 (74) Representative: Vossius & Partner Siebertstrasse 4 (71) Applicant: Oncotherapy Science, Inc. 81675 München (DE) Kawasaki-shi Kanagawa 213-0012 (JP) Remarks: This application was filed on 23-09-2010 as a divisional application to the application mentioned under INID code 62. (54) Method for diagnosing non-small cell lung cancers (57) Disclosed are methods for detecting non-small cancerous tissues are provided. Also disclosed are meth- cell lung cancer using differentially expressed genes. ods of identifying compounds for treating and preventing Furthermore, novel human genes whose expression is non-small cell lung cancer. -

Congenital Cataracts Due to a Novel 2‑Bp Deletion in CRYBA1/A3
1614 MOLECULAR MEDICINE REPORTS 10: 1614-1618, 2014 Congenital cataracts due to a novel 2‑bp deletion in CRYBA1/A3 JING ZHANG1, YANHUA ZHANG1, FANG FANG1, WEIHONG MU1, NING ZHANG2, TONGSHUN XU3 and QINYING CAO1 1Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital; 2Department of Cardiology, The Second Hospital of Hebei Medical University; 3Department of Surgery, Shijiazhuang Obstetrics and Gynecology Hospital, Shijiazhuang, Hebei, P.R. China Received September 22, 2013; Accepted April 11, 2014 DOI: 10.3892/mmr.2014.2324 Abstract. Congenital cataracts, which are a clinically and located in the eye lens. The major human crystallins comprise genetically heterogeneous group of eye disorders, lead to 90% of protein in the mature lens and contain two different visual impairment and are a significant cause of blindness superfamilies: the small heat‑shock proteins (α-crystallins) in childhood. A major proportion of the causative mutations and the βγ-crystallins. for congenital cataracts are found in crystallin genes. In the In this study a functional candidate approach was used present study, a novel deletion mutation (c.590-591delAG) in to investigate the known crystallin genes, including CRYAA, exon 6 of CRYBA1/A3 was identified in a large family with CRYAB, CRYBA1/A3, CRYBB1, CRYBB2, CRYGC, CRYGD autosomal dominant congenital cataracts. An increase in and CRYGS, in which a major proportion of the mutations local hydrophobicity was predicted around the mutation site; identified in a large family with congenital cataracts were however, further studies are required to determine the exact found. effect of the mutation on βA1/A3-crystallin structure and function. To the best of our knowledge, this is the first report Subjects and methods of an association between a frameshift mutation in exon 6 of CRYBA1/A3 and congenital cataracts. -

Perkinelmer Genomics to Request the Saliva Swab Collection Kit for Patients That Cannot Provide a Blood Sample As Whole Blood Is the Preferred Sample
Microphthalmia/ Anophthalmia/ Coloboma Spectrum Panel Test Code D4300 Test Summary This test analyzes 48 genes that have been associated with microphthalmia, anophthalmia, coloboma and/or disorders associated with these ocular findings Turn-Around-Time (TAT)* 3 - 5 weeks Acceptable Sample Types Whole Blood (EDTA) (Preferred sample type) DNA, Isolated Dried Blood Spots Saliva Acceptable Billing Types Self (patient) Payment Institutional Billing Commercial Insurance Indications for Testing Individual born with small eyes, missing a piece of their eye or missing their entire eye Individual suspected of having a syndrome associated with microphtalmia, anophthalmia, and/or colobomas Family planning purposes for individuals with a personal and/or family history of microphthalmia, anophthalmia, and/or coloboma Test Description This panel analyzes 48 genes that have been associated with microphthalmia, anophthalmia, coloboma and/or disorders associated with these ocular findings. Both sequencing and deletion/duplication (CNV) analysis will be performed on the coding regions of all genes included (unless otherwise marked). All analysis is performed utilizing Next Generation Sequencing (NGS) technology. CNV analysis is designed to detect the majority of deletions and duplications of three exons or greater in size. Smaller CNV events may also be detected and reported, but additional follow-up testing is recommended if a smaller CNV is suspected. All variants are classified according to ACMG guidelines. Condition Description Microphthalmia is an eye disease where an individual is born with one or both eyes being abnormally small. Anophthalmia is an eye disease where an individual is born without one or both eyes. A coloboma is an eye disease where an individual is born with a piece of their eye missing.