The Metrical Matrix in Teaching Mineralogy G. V
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Crystal Structure of a Material Is Way in Which Atoms, Ions, Molecules Are Spatially Arranged in 3-D Space
Crystalline Structures – The Basics •Crystal structure of a material is way in which atoms, ions, molecules are spatially arranged in 3-D space. •Crystal structure = lattice (unit cell geometry) + basis (atom, ion, or molecule positions placed on lattice points within the unit cell). •A lattice is used in context when describing crystalline structures, means a 3-D array of points in space. Every lattice point must have identical surroundings. •Unit cell: smallest repetitive volume •Each crystal structure is built by stacking which contains the complete lattice unit cells and placing objects (motifs, pattern of a crystal. A unit cell is chosen basis) on the lattice points: to represent the highest level of geometric symmetry of the crystal structure. It’s the basic structural unit or building block of crystal structure. 7 crystal systems in 3-D 14 crystal lattices in 3-D a, b, and c are the lattice constants 1 a, b, g are the interaxial angles Metallic Crystal Structures (the simplest) •Recall, that a) coulombic attraction between delocalized valence electrons and positively charged cores is isotropic (non-directional), b) typically, only one element is present, so all atomic radii are the same, c) nearest neighbor distances tend to be small, and d) electron cloud shields cores from each other. •For these reasons, metallic bonding leads to close packed, dense crystal structures that maximize space filling and coordination number (number of nearest neighbors). •Most elemental metals crystallize in the FCC (face-centered cubic), BCC (body-centered cubic, or HCP (hexagonal close packed) structures: Room temperature crystal structure Crystal structure just before it melts 2 Recall: Simple Cubic (SC) Structure • Rare due to low packing density (only a-Po has this structure) • Close-packed directions are cube edges. -

Types of Lattices
Types of Lattices Types of Lattices Lattices are either: 1. Primitive (or Simple): one lattice point per unit cell. 2. Non-primitive, (or Multiple) e.g. double, triple, etc.: more than one lattice point per unit cell. Double r2 cell r1 r2 Triple r1 cell r2 r1 Primitive cell N + e 4 Ne = number of lattice points on cell edges (shared by 4 cells) •When repeated by successive translations e =edge reproduce periodic pattern. •Multiple cells are usually selected to make obvious the higher symmetry (usually rotational symmetry) that is possessed by the 1 lattice, which may not be immediately evident from primitive cell. Lattice Points- Review 2 Arrangement of Lattice Points 3 Arrangement of Lattice Points (continued) •These are known as the basis vectors, which we will come back to. •These are not translation vectors (R) since they have non- integer values. The complexity of the system depends upon the symmetry requirements (is it lost or maintained?) by applying the symmetry operations (rotation, reflection, inversion and translation). 4 The Five 2-D Bravais Lattices •From the previous definitions of the four 2-D and seven 3-D crystal systems, we know that there are four and seven primitive unit cells (with 1 lattice point/unit cell), respectively. •We can then ask: can we add additional lattice points to the primitive lattices (or nets), in such a way that we still have a lattice (net) belonging to the same crystal system (with symmetry requirements)? •First illustrate this for 2-D nets, where we know that the surroundings of each lattice point must be identical. -
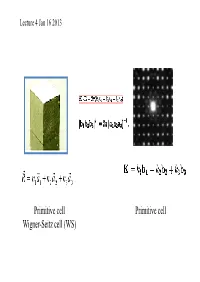
Primitive Cell Wigner-Seitz Cell (WS) Primitive Cell
Lecture 4 Jan 16 2013 Primitive cell Primitive cell Wigner-Seitz cell (WS) First Brillouin zone The Wigner-Seitz primitive cell of the reciprocal lattice is known as the first Brillouin zone. (Wigner-Seitz is real space concept while Brillouin zone is a reciprocal space idea). Powder cell Polymorphic Forms of Carbon Graphite – a soft, black, flaky solid, with a layered structure – parallel hexagonal arrays of carbon atoms – weak van der Waal’s forces between layers – planes slide easily over one another Miller indices Simple cubic Miller Indices Rules for determining Miller Indices: 1. Determine the intercepts of the face along the crystallographic axes, in terms of un it ce ll dimens ions. 2. Take the reciprocals 3. Clear fractions 4. Reduce to lowest terms Simple cubic d100=? Where does a protein crystallographer see the Miller indices? CtlCommon crystal faces are parallel to lattice planes • Eac h diffrac tion spo t can be regarded as a X-ray beam reflected from a lattice plane , and therefore has a unique Miller index. Miller indices A Miller index is a series of coprime integers that are inversely ppproportional to the intercepts of the cry stal face or crystallographic planes with the edges of the unit cell. It describes the orientation of a plane in the 3-D lattice with respect to the axes. The general form of the Miller index is (h, k, l) where h, k, and l are integers related to the unit cell along the a, b, c crystal axes. Irreducible brillouin zone II II II II Reciprocal lattice ghakblc The Bravais lattice after Fourier transform real space reciprocal lattice normaltthlls to the planes (vect ors ) poitints spacing between planes 1/distance between points ((y,p)actually, 2p/distance) l (distance, wavelength) 2p/l=k (momentum, wave number) BillBravais cell Wigner-SiSeitz ce ll Brillouin zone . -

Chapter 4, Bravais Lattice Primitive Vectors
Chapter 4, Bravais Lattice A Bravais lattice is the collection of all (and only those) points in space reachable from the origin with position vectors: n , n , n integer (+, -, or 0) r r r r 1 2 3 R = n1a1 + n2 a2 + n3a3 a1, a2, and a3 not all in same plane The three primitive vectors, a1, a2, and a3, uniquely define a Bravais lattice. However, for one Bravais lattice, there are many choices for the primitive vectors. A Bravais lattice is infinite. It is identical (in every aspect) when viewed from any of its lattice points. This is not a Bravais lattice. Honeycomb: P and Q are equivalent. R is not. A Bravais lattice can be defined as either the collection of lattice points, or the primitive translation vectors which construct the lattice. POINT Q OBJECT: Remember that a Bravais lattice has only points. Points, being dimensionless and isotropic, have full spatial symmetry (invariant under any point symmetry operation). Primitive Vectors There are many choices for the primitive vectors of a Bravais lattice. One sure way to find a set of primitive vectors (as described in Problem 4 .8) is the following: (1) a1 is the vector to a nearest neighbor lattice point. (2) a2 is the vector to a lattice points closest to, but not on, the a1 axis. (3) a3 is the vector to a lattice point nearest, but not on, the a18a2 plane. How does one prove that this is a set of primitive vectors? Hint: there should be no lattice points inside, or on the faces (lll)fhlhd(lllid)fd(parallolegrams) of, the polyhedron (parallelepiped) formed by these three vectors. -

Symmetry and Groups, and Crystal Structures
CHAPTER 3: SYMMETRY AND GROUPS, AND CRYSTAL STRUCTURES Sarah Lambart RECAP CHAP. 2 2 different types of close packing: hcp: tetrahedral interstice (ABABA) ccp: octahedral interstice (ABCABC) Definitions: The coordination number or CN is the number of closest neighbors of opposite charge around an ion. It can range from 2 to 12 in ionic structures. These structures are called coordination polyhedron. RECAP CHAP. 2 Rx/Rz C.N. Type Hexagonal or An ideal close-packing of sphere 1.0 12 Cubic for a given CN, can only be Closest Packing achieved for a specific ratio of 1.0 - 0.732 8 Cubic ionic radii between the anions and 0.732 - 0.414 6 Octahedral Tetrahedral (ex.: the cations. 0.414 - 0.225 4 4- SiO4 ) 0.225 - 0.155 3 Triangular <0.155 2 Linear RECAP CHAP. 2 Pauling’s rule: #1: the coodination polyhedron is defined by the ratio Rcation/Ranion #2: The Electrostatic Valency (e.v.) Principle: ev = Z/CN #3: Shared edges and faces of coordination polyhedra decreases the stability of the crystal. #4: In crystal with different cations, those of high valency and small CN tend not to share polyhedral elements #5: The principle of parsimony: The number of different sites in a crystal tends to be small. CONTENT CHAP. 3 (2-3 LECTURES) Definitions: unit cell and lattice 7 Crystal systems 14 Bravais lattices Element of symmetry CRYSTAL LATTICE IN TWO DIMENSIONS A crystal consists of atoms, molecules, or ions in a pattern that repeats in three dimensions. The geometry of the repeating pattern of a crystal can be described in terms of a crystal lattice, constructed by connecting equivalent points throughout the crystal. -

PHZ6426 Crystal Structure
SYMMETRY Finite geometric objects (“molecules) are symmetric with respect to a) rotations b) reflections (symmetry planes, symmetry points) c) combinatons of translations and rotations (screw axes) 2π If an object is symmetric with respect to rotation by angle φ = , it is said to have has an “n-fold rotational axis” n n=1 is a trivial case: every object is symmetric about a full revolution. Non-trivial axes have n>1. 1 Symmetries of an equilateral triangle 2 2 2 180-rotations (flips) 3’ 1’ about 22’ 3 1 3 1 2’ about 33’ about 11’ 1 3 1 2 2 3 3 2 2π 1 120 degree= rotations 3 3 2 1 3 3 3 4 2 2 1 1 An equilateral triangle is not symmetric with respect to inversion about its center. But a square is. 5 Gliding (screw) axis Rotate by 180 degrees about the axis Slide by a half-period 6 2-fold rotations can be viewed as reflections in a plane which contains a 2-fold rotation axis 7 Crystal Structure Crystal structure can be obtained by attaching atoms or groups of atoms --basis-- to lattice sites. Crystal Structure = Crystal Lattice + Basis Crystal Structure 88 Partially from Prof. C. W. Myles (Texas Tech) course presentation Building blocks of lattices are geometric figures with certain symmetries: rotational, reflectional, etc. Lattice must obey all these symmetries but, in addition, it also must obey TRANSLATIONAL symmetry Which geometric figures can be used to built a lattice? 9 10 Crystallographic Restriction Theorem: Crystal lattices have only 2-,3, 4-, and 6-fold rotational symmetries 11 B’ ma A’ n-fold axis φ-π/2 φ=2π/n a A B 1. -

Lattice Vibrations - Phonons
Lattice vibrations - phonons So far, we have assumed that the ions are fixed at their equilibrium positions, and we focussed on understanding the motion of the electrons in the static periodic potential created by the ions. But, of course, the ions are quantum objects that cannot be at rest in well-defined positions { this would violate Heisenberg's uncertainty principle. So they must be moving! In the following, we will assume that we are at temperatures very well below the melting temperature of the crystal, so that the ions perform small oscillations about their equilibrium positions. Of course, with increasing T the amplitude of these vibrations increases and eventually becomes large enough that the ions start moving past one another and the lattice order is progressively lost (the crystal starts to melt); our approximations will not be valid once we reach such high temperatures. Dealing with small oscillations is very nice (as you may remember from studying Lagrangian/Hamiltonian dynamics) because in the harmonic approximation, i.e. where we expand potentials up to second- order in these displacements, such classical problems can be solved exactly by mapping them to a sum of independent harmonic oscillators. Going to the quantum solution is then trivial, because it will simply be the sum of independent quantum harmonic oscillators. In this chapter we dis- cuss this solution and its implications, and for the time being we will completely forget about the electrons. We will return back to them in the next section. I assume you have already studied phonons in your undergraduate solid state physics course. -

Crystal Structure
Physics 927 E.Y.Tsymbal Section 1: Crystal Structure A solid is said to be a crystal if atoms are arranged in such a way that their positions are exactly periodic. This concept is illustrated in Fig.1 using a two-dimensional (2D) structure. y T C Fig.1 A B a x 1 A perfect crystal maintains this periodicity in both the x and y directions from -∞ to +∞. As follows from this periodicity, the atoms A, B, C, etc. are equivalent. In other words, for an observer located at any of these atomic sites, the crystal appears exactly the same. The same idea can be expressed by saying that a crystal possesses a translational symmetry. The translational symmetry means that if the crystal is translated by any vector joining two atoms, say T in Fig.1, the crystal appears exactly the same as it did before the translation. In other words the crystal remains invariant under any such translation. The structure of all crystals can be described in terms of a lattice, with a group of atoms attached to every lattice point. For example, in the case of structure shown in Fig.1, if we replace each atom by a geometrical point located at the equilibrium position of that atom, we obtain a crystal lattice. The crystal lattice has the same geometrical properties as the crystal, but it is devoid of any physical contents. There are two classes of lattices: the Bravais and the non-Bravais. In a Bravais lattice all lattice points are equivalent and hence by necessity all atoms in the crystal are of the same kind. -

02 Geometry, Reclattice.Pdf
Geometry of Crystals Crystal is a solid composed of atoms, ions or molecules that demonstrate long range periodic order in three dimensions The Crystalline State State of Fixed Fixed Order Properties Matter Volume Shape Gas No No No Isotropic Liquid Yes No Short-range Isotropic Solid Yes Yes Short-range Isotropic (amorphous) Solid Yes Yes Long-range Anisotropic (crystalline) Atoms Lattice constants A Crystal Lattice a, b B C Not only atom, ion or molecule b positions are repetitious – there are a certain symmetry relationships in their arrangement. Crystalline Lattice structure = Basis + Crystal Lattice a a r ua One-dimensional lattice with lattice parameter a a a r ua b b b Two-dimensional lattice with lattice parameters a, b and Crystal Lattice r ua b wc Crystal Lattice Lattice vectors, lattice parameters and interaxial angles c Lattice vector a b c c Lattice parameter a b c b Interaxial angle b a a A lattice is an array of points in space in which the environment of each point is identical Crystal Lattice Lattice Not a lattice Crystal Lattice Unit cell content Coordinates of all atoms Types of atoms Site occupancy Individual displacement parameters y1 y2 y3 0 x1 x2 x3 Crystal Lattice Usually unit cell has more than one molecule or group of atoms They can be represented by symmetry operators rotation Symmetry Symmetry is a property of a crystal which is used to describe repetitions of a pattern within that crystal. Description is done using symmetry operators Rotation (about axis O) = 360°/n where n is the fold of the axis Translation n = 1, 2, 3, 4 or 6) m O i Mirror reflection Inversion Two-dimensional Symmetry Elements 1. -

MIT 8.231 Problem 2.1 Americium
MIT 8.231 Problem 2.1 Americium March 28, 2015 Problem The figure below shows a primitive unit cell of one crystalline form of the elementp americium. The space lattice is hexagonal with ~a1 = a~x;~a2 = 1=2a~x + 1=2 3a~y; and ~a3 = c~z. The basis is (0 0 0) , (2/3 2/3 1/4), (0 0 1/2) and (1/3 1/3 3/4). a) Find the reciprocal lattice vectors G~ . Describe in words and sketch the reciprocal lattice. b) Sketch the first Brillouin zone. Give values for the important dimensions. c) Find the structure factors associated with the points (100), (001), and (120) of the reciprocal lattice. d) What is the ratio of the scattering intensity corresponding to (120) to that corresponding to (100)? 1 Solution a) ~ ~ ~ At first we want to calculate reciprocal lattice vectors b1; b2; b3. The vectors are defined as followed: ~ a2 × a3 b1 = 2π (1) a1(a2 × a3) ~ a3 × a1 b2 = 2π (2) a1(a2 × a3) ~ a1 × a2 b3 = 2π (3) a1(a2 × a3) With the given values of ~ai we get: p 0 31 ~ 2π b1 = p @−1A (4) 3a 0 001 ~ 4π b2 = p @1A (5) 3a 0 001 2π b~ = 0 (6) 3 c @ A 1 The reciprocal lattice vector is defined as followed: ~ ~ ~ G~ = hb1 + kb2 + lb3 (7) Where h,k,l are Miller indices, with those indices Crystal planes are defined. G~ is also used for x-ray diffraction, the condition for diffraction is defined as: ~ ~ ~ G~ hkl = ∆k = k − k0 (8) This is best shown with the Ewald's Sphere. -

Introduction to Solid State Physics
Introduction to Solid state physics Chapter 1 Crystal Structures 晶體結構 Crystal Structures Periodic arrays of atoms Fundamental types of lattices Index system for crystal plans Simple crystal structures Direct image of atomic structure Non-ideal crystal structures Introduction In 1895, a German physicist, W. C. Roentgen discovered x-ray. In 1912 Laue developed an elementary theory of the diffraction of x-rays by a periodic array. In the second part, Friedrich and Knipping reported the first experimental observations of x-ray diffraction by crystals. 2 The work proved decisively that crystals are composed of a periodic array of atoms. The studies have been extended to include amorphous or noncrystalline solids, glasses, and liquids. The wider field is known as condensed matter physics. Periodic Arrays of Atoms An ideal crystal is constructed by the infinite repetition of identical structural units in space. The structural unit is a single atom, comprise many atoms or molecules. 晶格 The structure of all crystals can be described in terms of a lattice, with a group of atoms attached to every lattice point. The group of atoms is called the basis. 基底 The concepts of Lattice & Basis . x-ray diffraction . neutron diffraction . electron diffraction 晶格 + 基底 = 晶體結構 With this definition of the primitive translation vectors, there is no cell of smaller volume that can serve as a building block for the crystal structure. The crystal axes a1, a2, a3 form three adjacent edges of a parallelepiped. If there are lattice points only at the corners, then it is a primitive parallelepiped. Primitive Lattice Cell The parallelepiped defined by primitive axes a1, a2, a3 is called a primitive cell (Fig. -

Crystal Structure CRYSTAL STRUCTURES
Crystal Structure CRYSTAL STRUCTURES Lecture 2 A.H. Harker Physics and Astronomy UCL Lattice and Basis Version: June 6, 2002; Typeset on October 30, 2003,15:28 2 1.4.8 Fourteen Lattices in Three Dimensions Version: June 6, 2002; Typeset on October 30, 2003,15:28 3 System Type Restrictions Triclinic P a =6 b =6 c, α =6 β =6 γ Monoclinic P,C a =6 b =6 c, α = γ = 90◦ =6 β Orthorhmobic P,C,I,F a =6 b =6 c, α = β = γ = 90◦ Tetragonal P,I a = b =6 c, α = β = γ = 90◦ Cubic P,I,F1 a = b = c, α = β = γ = 90◦ Trigonal P a = b = c, α = β = γ < 120◦, =6 90◦ Hexagonal P a = b =6 c, α = β = 90◦, γ = 120◦ No need to learn details except for cubic, basic ideas of hexagonal. Version: June 6, 2002; Typeset on October 30, 2003,15:28 4 1.4.9 Cubic Unit Cells Primitive Unit Cells of cubic system Simple cubic: cube containing one lattice point (or 8 corner points each shared among 8 cubes: 8 × 1 8 = 1). Body centred cubic: 2 points in cubic cell Version: June 6, 2002; Typeset on October 30, 2003,15:28 5 Rhombohedral primitive cell of body centred cubic system. Face centred cubic: 4 points in cubic cell (8 corner points shared 8 1 1 ways, 6 face points shared 2 ways: 8 × 8 + 6 × 2 = 4. Version: June 6, 2002; Typeset on October 30, 2003,15:28 6 Rhombohedral primitive cell of face centred cubic system.