Diamond Unit Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Crystal Structures
Crystal Structures Academic Resource Center Crystallinity: Repeating or periodic array over large atomic distances. 3-D pattern in which each atom is bonded to its nearest neighbors Crystal structure: the manner in which atoms, ions, or molecules are spatially arranged. Unit cell: small repeating entity of the atomic structure. The basic building block of the crystal structure. It defines the entire crystal structure with the atom positions within. Lattice: 3D array of points coinciding with atom positions (center of spheres) Metallic Crystal Structures FCC (face centered cubic): Atoms are arranged at the corners and center of each cube face of the cell. FCC continued Close packed Plane: On each face of the cube Atoms are assumed to touch along face diagonals. 4 atoms in one unit cell. a 2R 2 BCC: Body Centered Cubic • Atoms are arranged at the corners of the cube with another atom at the cube center. BCC continued • Close Packed Plane cuts the unit cube in half diagonally • 2 atoms in one unit cell 4R a 3 Hexagonal Close Packed (HCP) • Cell of an HCP lattice is visualized as a top and bottom plane of 7 atoms, forming a regular hexagon around a central atom. In between these planes is a half- hexagon of 3 atoms. • There are two lattice parameters in HCP, a and c, representing the basal and height parameters Volume respectively. 6 atoms per unit cell Coordination number – the number of nearest neighbor atoms or ions surrounding an atom or ion. For FCC and HCP systems, the coordination number is 12. For BCC it’s 8. -

361: Crystallography and Diffraction
361: Crystallography and Diffraction Michael Bedzyk Department of Materials Science and Engineering Northwestern University February 2, 2021 Contents 1 Catalog Description3 2 Course Outcomes3 3 361: Crystallography and Diffraction3 4 Symmetry of Crystals4 4.1 Types of Symmetry.......................... 4 4.2 Projections of Symmetry Elements and Point Groups...... 9 4.3 Translational Symmetry ....................... 17 5 Crystal Lattices 18 5.1 Indexing within a crystal lattice................... 18 5.2 Lattices................................. 22 6 Stereographic Projections 31 6.1 Projection plane............................ 33 6.2 Point of projection........................... 33 6.3 Representing Atomic Planes with Vectors............. 36 6.4 Concept of Reciprocal Lattice .................... 38 6.5 Reciprocal Lattice Vector....................... 41 7 Representative Crystal Structures 48 7.1 Crystal Structure Examples ..................... 48 7.2 The hexagonal close packed structure, unlike the examples in Figures 7.1 and 7.2, has two atoms per lattice point. These two atoms are considered to be the motif, or repeating object within the HCP lattice. ............................ 49 1 7.3 Voids in FCC.............................. 54 7.4 Atom Sizes and Coordination.................... 54 8 Introduction to Diffraction 57 8.1 X-ray .................................. 57 8.2 Interference .............................. 57 8.3 X-ray Diffraction History ...................... 59 8.4 How does X-ray diffraction work? ................. 59 8.5 Absent -

Crystal Structure of a Material Is Way in Which Atoms, Ions, Molecules Are Spatially Arranged in 3-D Space
Crystalline Structures – The Basics •Crystal structure of a material is way in which atoms, ions, molecules are spatially arranged in 3-D space. •Crystal structure = lattice (unit cell geometry) + basis (atom, ion, or molecule positions placed on lattice points within the unit cell). •A lattice is used in context when describing crystalline structures, means a 3-D array of points in space. Every lattice point must have identical surroundings. •Unit cell: smallest repetitive volume •Each crystal structure is built by stacking which contains the complete lattice unit cells and placing objects (motifs, pattern of a crystal. A unit cell is chosen basis) on the lattice points: to represent the highest level of geometric symmetry of the crystal structure. It’s the basic structural unit or building block of crystal structure. 7 crystal systems in 3-D 14 crystal lattices in 3-D a, b, and c are the lattice constants 1 a, b, g are the interaxial angles Metallic Crystal Structures (the simplest) •Recall, that a) coulombic attraction between delocalized valence electrons and positively charged cores is isotropic (non-directional), b) typically, only one element is present, so all atomic radii are the same, c) nearest neighbor distances tend to be small, and d) electron cloud shields cores from each other. •For these reasons, metallic bonding leads to close packed, dense crystal structures that maximize space filling and coordination number (number of nearest neighbors). •Most elemental metals crystallize in the FCC (face-centered cubic), BCC (body-centered cubic, or HCP (hexagonal close packed) structures: Room temperature crystal structure Crystal structure just before it melts 2 Recall: Simple Cubic (SC) Structure • Rare due to low packing density (only a-Po has this structure) • Close-packed directions are cube edges. -

Types of Lattices
Types of Lattices Types of Lattices Lattices are either: 1. Primitive (or Simple): one lattice point per unit cell. 2. Non-primitive, (or Multiple) e.g. double, triple, etc.: more than one lattice point per unit cell. Double r2 cell r1 r2 Triple r1 cell r2 r1 Primitive cell N + e 4 Ne = number of lattice points on cell edges (shared by 4 cells) •When repeated by successive translations e =edge reproduce periodic pattern. •Multiple cells are usually selected to make obvious the higher symmetry (usually rotational symmetry) that is possessed by the 1 lattice, which may not be immediately evident from primitive cell. Lattice Points- Review 2 Arrangement of Lattice Points 3 Arrangement of Lattice Points (continued) •These are known as the basis vectors, which we will come back to. •These are not translation vectors (R) since they have non- integer values. The complexity of the system depends upon the symmetry requirements (is it lost or maintained?) by applying the symmetry operations (rotation, reflection, inversion and translation). 4 The Five 2-D Bravais Lattices •From the previous definitions of the four 2-D and seven 3-D crystal systems, we know that there are four and seven primitive unit cells (with 1 lattice point/unit cell), respectively. •We can then ask: can we add additional lattice points to the primitive lattices (or nets), in such a way that we still have a lattice (net) belonging to the same crystal system (with symmetry requirements)? •First illustrate this for 2-D nets, where we know that the surroundings of each lattice point must be identical. -

The Metrical Matrix in Teaching Mineralogy G. V
THE METRICAL MATRIX IN TEACHING MINERALOGY G. V. GIBBS Department of Geological Sciences & Department of Materials Science and Engineering Virginia Polytechnic Institute and State University Blacksburg, VA 24061 [email protected] INTRODUCTION The calculation of the d-spacings, the angles between planes and zones, the bond lengths and angles and other important geometric relationships for a mineral can be a tedious task both for the student and the instructor, particularly when completed with the large as- sortment of trigonometric identities and algebraic formulae that are available (d. Crystal Geometry (1959), Donnay and Donnay, International Tables for Crystallography, Vol. II, Section 3, The Kynoch Press, 101-158). However, such calculations are straightforward and relatively easy to do when completed with the metrical matrix and the interactive software MATOP. Several applications of the matrix are presented below, each of which is worked out in detail and which is designed to teach you its use in the study of crystal geometry. SOME PRELIMINARY COMMENTS We begin our discussion of the matrix with a brief examination of the properties of the geometric three dimensional space, S, in which we live and in which minerals and rocks occur. For our purposes, it will be convenient to view S as the set of all vectors that radiate from a common origin to each point in space. In constructing a model for S, we chose three noncoplanar, coordinate axes denoted X, Y and Z, each radiating from the origin, O. Next, we place three nonzero vectors denoted a, band c along X, Y and Z, respectively, likewise radiating from O. -
Primitive Cell Wigner-Seitz Cell (WS) Primitive Cell
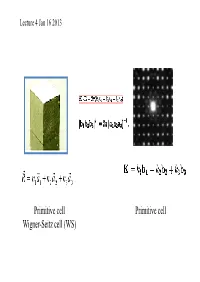
Lecture 4 Jan 16 2013 Primitive cell Primitive cell Wigner-Seitz cell (WS) First Brillouin zone The Wigner-Seitz primitive cell of the reciprocal lattice is known as the first Brillouin zone. (Wigner-Seitz is real space concept while Brillouin zone is a reciprocal space idea). Powder cell Polymorphic Forms of Carbon Graphite – a soft, black, flaky solid, with a layered structure – parallel hexagonal arrays of carbon atoms – weak van der Waal’s forces between layers – planes slide easily over one another Miller indices Simple cubic Miller Indices Rules for determining Miller Indices: 1. Determine the intercepts of the face along the crystallographic axes, in terms of un it ce ll dimens ions. 2. Take the reciprocals 3. Clear fractions 4. Reduce to lowest terms Simple cubic d100=? Where does a protein crystallographer see the Miller indices? CtlCommon crystal faces are parallel to lattice planes • Eac h diffrac tion spo t can be regarded as a X-ray beam reflected from a lattice plane , and therefore has a unique Miller index. Miller indices A Miller index is a series of coprime integers that are inversely ppproportional to the intercepts of the cry stal face or crystallographic planes with the edges of the unit cell. It describes the orientation of a plane in the 3-D lattice with respect to the axes. The general form of the Miller index is (h, k, l) where h, k, and l are integers related to the unit cell along the a, b, c crystal axes. Irreducible brillouin zone II II II II Reciprocal lattice ghakblc The Bravais lattice after Fourier transform real space reciprocal lattice normaltthlls to the planes (vect ors ) poitints spacing between planes 1/distance between points ((y,p)actually, 2p/distance) l (distance, wavelength) 2p/l=k (momentum, wave number) BillBravais cell Wigner-SiSeitz ce ll Brillouin zone . -

Chapter 4, Bravais Lattice Primitive Vectors
Chapter 4, Bravais Lattice A Bravais lattice is the collection of all (and only those) points in space reachable from the origin with position vectors: n , n , n integer (+, -, or 0) r r r r 1 2 3 R = n1a1 + n2 a2 + n3a3 a1, a2, and a3 not all in same plane The three primitive vectors, a1, a2, and a3, uniquely define a Bravais lattice. However, for one Bravais lattice, there are many choices for the primitive vectors. A Bravais lattice is infinite. It is identical (in every aspect) when viewed from any of its lattice points. This is not a Bravais lattice. Honeycomb: P and Q are equivalent. R is not. A Bravais lattice can be defined as either the collection of lattice points, or the primitive translation vectors which construct the lattice. POINT Q OBJECT: Remember that a Bravais lattice has only points. Points, being dimensionless and isotropic, have full spatial symmetry (invariant under any point symmetry operation). Primitive Vectors There are many choices for the primitive vectors of a Bravais lattice. One sure way to find a set of primitive vectors (as described in Problem 4 .8) is the following: (1) a1 is the vector to a nearest neighbor lattice point. (2) a2 is the vector to a lattice points closest to, but not on, the a1 axis. (3) a3 is the vector to a lattice point nearest, but not on, the a18a2 plane. How does one prove that this is a set of primitive vectors? Hint: there should be no lattice points inside, or on the faces (lll)fhlhd(lllid)fd(parallolegrams) of, the polyhedron (parallelepiped) formed by these three vectors. -

Multidisciplinary Design Project Engineering Dictionary Version 0.0.2
Multidisciplinary Design Project Engineering Dictionary Version 0.0.2 February 15, 2006 . DRAFT Cambridge-MIT Institute Multidisciplinary Design Project This Dictionary/Glossary of Engineering terms has been compiled to compliment the work developed as part of the Multi-disciplinary Design Project (MDP), which is a programme to develop teaching material and kits to aid the running of mechtronics projects in Universities and Schools. The project is being carried out with support from the Cambridge-MIT Institute undergraduate teaching programe. For more information about the project please visit the MDP website at http://www-mdp.eng.cam.ac.uk or contact Dr. Peter Long Prof. Alex Slocum Cambridge University Engineering Department Massachusetts Institute of Technology Trumpington Street, 77 Massachusetts Ave. Cambridge. Cambridge MA 02139-4307 CB2 1PZ. USA e-mail: [email protected] e-mail: [email protected] tel: +44 (0) 1223 332779 tel: +1 617 253 0012 For information about the CMI initiative please see Cambridge-MIT Institute website :- http://www.cambridge-mit.org CMI CMI, University of Cambridge Massachusetts Institute of Technology 10 Miller’s Yard, 77 Massachusetts Ave. Mill Lane, Cambridge MA 02139-4307 Cambridge. CB2 1RQ. USA tel: +44 (0) 1223 327207 tel. +1 617 253 7732 fax: +44 (0) 1223 765891 fax. +1 617 258 8539 . DRAFT 2 CMI-MDP Programme 1 Introduction This dictionary/glossary has not been developed as a definative work but as a useful reference book for engi- neering students to search when looking for the meaning of a word/phrase. It has been compiled from a number of existing glossaries together with a number of local additions. -

Theoretical Study of Ternary Cosp Semiconductor
Theoretical Study of Ternary CoSP Semiconductor: a Candidate for Photovoltaic Applications Abdesalem Houari∗ and Fares Benissad Theoretical Physics Laboratory, Department of Physics, University of Bejaia, Bejaia, Algeria (Dated: March 24, 2020) Abstract The electronic structure of pyrite-type cobalt phosphosulfide (CoSP) has been studied using density-functional theory. The calculated band structure reveals the non-magnetic semiconducting character of the compound. The electronic structure is described through the electronic band structure and the densities of states. A band gap of 1.14 eV has been computed within standard GGA, a value which is enhanced using hybrid functional. It separates the upper part of the valence band dominated by Co-3d-t2g states from the lower part of the conduction band made exclusively of Co-3d-eg, above of which lie S-3p and P-3p ones. The obtained values are suitable for applications in solar cells, according to Shockley-Queisser theory of light to electric conversion efficiency. The origin of the larger CoSP band gap, with respect to the one of the promising FeS2 compound, is explained and the chemical bonding properties are addressed. A comparative picture is established where several similarities have been found, suggesting that CoSP could be for a great practical interest in photovoltaics. arXiv:2003.09864v1 [cond-mat.mtrl-sci] 22 Mar 2020 ∗corresponding author:[email protected] 1 Contents I. Introduction 2 II. Computational Method 4 III. Results and Discussions 5 IV. Summary and Conclusion 9 Acknowledgments 10 References 10 I. INTRODUCTION Pyrite-type iron disulfide (FeS2) is considered as a promising material for photovoltaic applications [1–3]. -

Crystal Structure
Physics 927 E.Y.Tsymbal Section 1: Crystal Structure A solid is said to be a crystal if atoms are arranged in such a way that their positions are exactly periodic. This concept is illustrated in Fig.1 using a two-dimensional (2D) structure. y T C Fig.1 A B a x 1 A perfect crystal maintains this periodicity in both the x and y directions from -∞ to +∞. As follows from this periodicity, the atoms A, B, C, etc. are equivalent. In other words, for an observer located at any of these atomic sites, the crystal appears exactly the same. The same idea can be expressed by saying that a crystal possesses a translational symmetry. The translational symmetry means that if the crystal is translated by any vector joining two atoms, say T in Fig.1, the crystal appears exactly the same as it did before the translation. In other words the crystal remains invariant under any such translation. The structure of all crystals can be described in terms of a lattice, with a group of atoms attached to every lattice point. For example, in the case of structure shown in Fig.1, if we replace each atom by a geometrical point located at the equilibrium position of that atom, we obtain a crystal lattice. The crystal lattice has the same geometrical properties as the crystal, but it is devoid of any physical contents. There are two classes of lattices: the Bravais and the non-Bravais. In a Bravais lattice all lattice points are equivalent and hence by necessity all atoms in the crystal are of the same kind. -

The American Mineralogist Crystal Structure Database
American Mineralogist, Volume 88, pages 247–250, 2003 The American Mineralogist crystal structure database ROBERT T. DOWNS* AND MICHELLE HALL-WALLACE Department of Geosciences, University of Arizona, Tucson, Arizona 85721-0077, U.S.A. ABSTRACT A database has been constructed that contains all the crystal structures previously published in the American Mineralogist. The database is called “The American Mineralogist Crystal Structure Database” and is freely accessible from the websites of the Mineralogical Society of America at http://www.minsocam.org/MSA/Crystal_Database.html and the University of Arizona. In addition to the database, a suite of interactive software is provided that can be used to view and manipulate the crystal structures and compute different properties of a crystal such as geometry, diffraction patterns, and procrystal electron densities. The database is set up so that the data can be easily incorporated into other software packages. Included at the website is an evolving set of guides to instruct the user and help with classroom education. INTRODUCTION parameters; (5) incorporating comments from either the origi- The structure of a crystal represents a minimum energy con- nal authors or ourselves when changes are made to the origi- figuration adopted by a collection of atoms at a given tempera- nally published data. Each record in the database consists of a ture and pressure. In principle, all the physical and chemical bibliographic reference, cell parameters, symmetry, atomic properties of any crystalline substance can be computed from positions, displacement parameters, and site occupancies. An knowledge of its crystal structure. The determination of crys- example of a data set is provided in Figure 1. -

Crystals the Atoms in a Crystal Are Arranged in a Periodic Pattern. A
Crystals The atoms in a crystal are arranged in a periodic pattern. A crystal is made up of unit cells that are repeated at a pattern of points, which is called the Bravais lattice (fig. (1)). The Bravais lattice is basically a three dimensional abstract lattice of points. Figure 1: Unit cell - Bravais lattice - Crystal Unit cell The unit cell is the description of the arrangement of the atoms that get repeated, and is not unique - there are a various possibilities to choose it. The primitive unit cell is the smallest possible pattern to reproduce the crystal and is also not unique. As shown in fig. (2) for 2 dimensions there is a number of options of choosing the primitive unit cell. Nevertheless all have the same area. The same is true in three dimensions (various ways to choose primitive unit cell; same volume). An example for a possible three dimensional primitive unit cell and the formula for calculating the volume can be seen in fig. (2). Figure 2: Illustration of primitive unit cells 1 The conventional unit cell is agreed upon. It is chosen in a way to display the crystal or the crystal symmetries as good as possible. Normally it is easier to imagine things when looking at the conventional unit cell. A few conventional unit cells are displayed in fig. (3). Figure 3: Illustration of conventional unit cells • sc (simple cubic): Conventional and primitive unit cell are identical • fcc (face centered cubic): The conventional unit cell consists of 4 atoms in com- parison to the primitive unit cell which consists of only 1 atom.