Crystal Structure of a Material Is Way in Which Atoms, Ions, Molecules Are Spatially Arranged in 3-D Space
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Crystal Structures
Crystal Structures Academic Resource Center Crystallinity: Repeating or periodic array over large atomic distances. 3-D pattern in which each atom is bonded to its nearest neighbors Crystal structure: the manner in which atoms, ions, or molecules are spatially arranged. Unit cell: small repeating entity of the atomic structure. The basic building block of the crystal structure. It defines the entire crystal structure with the atom positions within. Lattice: 3D array of points coinciding with atom positions (center of spheres) Metallic Crystal Structures FCC (face centered cubic): Atoms are arranged at the corners and center of each cube face of the cell. FCC continued Close packed Plane: On each face of the cube Atoms are assumed to touch along face diagonals. 4 atoms in one unit cell. a 2R 2 BCC: Body Centered Cubic • Atoms are arranged at the corners of the cube with another atom at the cube center. BCC continued • Close Packed Plane cuts the unit cube in half diagonally • 2 atoms in one unit cell 4R a 3 Hexagonal Close Packed (HCP) • Cell of an HCP lattice is visualized as a top and bottom plane of 7 atoms, forming a regular hexagon around a central atom. In between these planes is a half- hexagon of 3 atoms. • There are two lattice parameters in HCP, a and c, representing the basal and height parameters Volume respectively. 6 atoms per unit cell Coordination number – the number of nearest neighbor atoms or ions surrounding an atom or ion. For FCC and HCP systems, the coordination number is 12. For BCC it’s 8. -

Tetrahedral Coordination with Lone Pairs
TETRAHEDRAL COORDINATION WITH LONE PAIRS In the examples we have discussed so far, the shape of the molecule is defined by the coordination geometry; thus the carbon in methane is tetrahedrally coordinated, and there is a hydrogen at each corner of the tetrahedron, so the molecular shape is also tetrahedral. It is common practice to represent bonding patterns by "generic" formulas such as AX4, AX2E2, etc., in which "X" stands for bonding pairs and "E" denotes lone pairs. (This convention is known as the "AXE method") The bonding geometry will not be tetrahedral when the valence shell of the central atom contains nonbonding electrons, however. The reason is that the nonbonding electrons are also in orbitals that occupy space and repel the other orbitals. This means that in figuring the coordination number around the central atom, we must count both the bonded atoms and the nonbonding pairs. The water molecule: AX2E2 In the water molecule, the central atom is O, and the Lewis electron dot formula predicts that there will be two pairs of nonbonding electrons. The oxygen atom will therefore be tetrahedrally coordinated, meaning that it sits at the center of the tetrahedron as shown below. Two of the coordination positions are occupied by the shared electron-pairs that constitute the O–H bonds, and the other two by the non-bonding pairs. Thus although the oxygen atom is tetrahedrally coordinated, the bonding geometry (shape) of the H2O molecule is described as bent. There is an important difference between bonding and non-bonding electron orbitals. Because a nonbonding orbital has no atomic nucleus at its far end to draw the electron cloud toward it, the charge in such an orbital will be concentrated closer to the central atom. -

Types of Lattices
Types of Lattices Types of Lattices Lattices are either: 1. Primitive (or Simple): one lattice point per unit cell. 2. Non-primitive, (or Multiple) e.g. double, triple, etc.: more than one lattice point per unit cell. Double r2 cell r1 r2 Triple r1 cell r2 r1 Primitive cell N + e 4 Ne = number of lattice points on cell edges (shared by 4 cells) •When repeated by successive translations e =edge reproduce periodic pattern. •Multiple cells are usually selected to make obvious the higher symmetry (usually rotational symmetry) that is possessed by the 1 lattice, which may not be immediately evident from primitive cell. Lattice Points- Review 2 Arrangement of Lattice Points 3 Arrangement of Lattice Points (continued) •These are known as the basis vectors, which we will come back to. •These are not translation vectors (R) since they have non- integer values. The complexity of the system depends upon the symmetry requirements (is it lost or maintained?) by applying the symmetry operations (rotation, reflection, inversion and translation). 4 The Five 2-D Bravais Lattices •From the previous definitions of the four 2-D and seven 3-D crystal systems, we know that there are four and seven primitive unit cells (with 1 lattice point/unit cell), respectively. •We can then ask: can we add additional lattice points to the primitive lattices (or nets), in such a way that we still have a lattice (net) belonging to the same crystal system (with symmetry requirements)? •First illustrate this for 2-D nets, where we know that the surroundings of each lattice point must be identical. -

The Metrical Matrix in Teaching Mineralogy G. V
THE METRICAL MATRIX IN TEACHING MINERALOGY G. V. GIBBS Department of Geological Sciences & Department of Materials Science and Engineering Virginia Polytechnic Institute and State University Blacksburg, VA 24061 [email protected] INTRODUCTION The calculation of the d-spacings, the angles between planes and zones, the bond lengths and angles and other important geometric relationships for a mineral can be a tedious task both for the student and the instructor, particularly when completed with the large as- sortment of trigonometric identities and algebraic formulae that are available (d. Crystal Geometry (1959), Donnay and Donnay, International Tables for Crystallography, Vol. II, Section 3, The Kynoch Press, 101-158). However, such calculations are straightforward and relatively easy to do when completed with the metrical matrix and the interactive software MATOP. Several applications of the matrix are presented below, each of which is worked out in detail and which is designed to teach you its use in the study of crystal geometry. SOME PRELIMINARY COMMENTS We begin our discussion of the matrix with a brief examination of the properties of the geometric three dimensional space, S, in which we live and in which minerals and rocks occur. For our purposes, it will be convenient to view S as the set of all vectors that radiate from a common origin to each point in space. In constructing a model for S, we chose three noncoplanar, coordinate axes denoted X, Y and Z, each radiating from the origin, O. Next, we place three nonzero vectors denoted a, band c along X, Y and Z, respectively, likewise radiating from O. -
Primitive Cell Wigner-Seitz Cell (WS) Primitive Cell
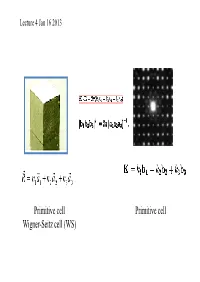
Lecture 4 Jan 16 2013 Primitive cell Primitive cell Wigner-Seitz cell (WS) First Brillouin zone The Wigner-Seitz primitive cell of the reciprocal lattice is known as the first Brillouin zone. (Wigner-Seitz is real space concept while Brillouin zone is a reciprocal space idea). Powder cell Polymorphic Forms of Carbon Graphite – a soft, black, flaky solid, with a layered structure – parallel hexagonal arrays of carbon atoms – weak van der Waal’s forces between layers – planes slide easily over one another Miller indices Simple cubic Miller Indices Rules for determining Miller Indices: 1. Determine the intercepts of the face along the crystallographic axes, in terms of un it ce ll dimens ions. 2. Take the reciprocals 3. Clear fractions 4. Reduce to lowest terms Simple cubic d100=? Where does a protein crystallographer see the Miller indices? CtlCommon crystal faces are parallel to lattice planes • Eac h diffrac tion spo t can be regarded as a X-ray beam reflected from a lattice plane , and therefore has a unique Miller index. Miller indices A Miller index is a series of coprime integers that are inversely ppproportional to the intercepts of the cry stal face or crystallographic planes with the edges of the unit cell. It describes the orientation of a plane in the 3-D lattice with respect to the axes. The general form of the Miller index is (h, k, l) where h, k, and l are integers related to the unit cell along the a, b, c crystal axes. Irreducible brillouin zone II II II II Reciprocal lattice ghakblc The Bravais lattice after Fourier transform real space reciprocal lattice normaltthlls to the planes (vect ors ) poitints spacing between planes 1/distance between points ((y,p)actually, 2p/distance) l (distance, wavelength) 2p/l=k (momentum, wave number) BillBravais cell Wigner-SiSeitz ce ll Brillouin zone . -

Oxide Crystal Structures: the Basics
Oxide crystal structures: The basics Ram Seshadri Materials Department and Department of Chemistry & Biochemistry Materials Research Laboratory, University of California, Santa Barbara CA 93106 USA [email protected] Originally created for the: ICMR mini-School at UCSB: Computational tools for functional oxide materials – An introduction for experimentalists This lecture 1. Brief description of oxide crystal structures (simple and complex) a. Ionic radii and Pauling’s rules b. Electrostatic valence c. Bond valence, and bond valence sums Why do certain combinations of atoms take on specific structures? My bookshelf H. D. Megaw O. Muller & R. Roy I. D. Brown B. G. Hyde & S. Andersson Software: ICSD + VESTA K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Cryst. 44 (2011) 1272–1276. [doi:10.1107/S0021889811038970] Crystal structures of simple oxides [containing a single cation site] Crystal structures of simple oxides [containing a single cation site] N.B.: CoO is simple, Co3O4 is not. ZnCo2O4 is certainly not ! Co3O4 and ZnCo2O4 are complex oxides. Graphs of connectivity in crystals: Atoms are nodes and edges (the lines that connect nodes) indicate short (near-neighbor) distances. CO2: The molecular structure is O=C=O. The graph is: Each C connected to 2 O, each O connected to a 1 C OsO4: The structure comprises isolated tetrahedra (molecular). The graph is below: Each Os connected to 4 O and each O to 1 Os Crystal structures of simple oxides of monovalent ions: A2O Cu2O Linear coordination is unusual. Found usually in Cu+ and Ag+. -

Inorganic Chemistry
Inorganic Chemistry Chemistry 120 Fall 2005 Prof. Seth Cohen Some Important Features of Metal Ions Electronic configuration • Oxidation State/Charge. •Size. • Coordination number. • Coordination geometry. • “Soft vs. Hard”. • Lability. • Electrochemistry. • Ligand environment. 1 Oxidation State/Charge • The oxidation state describes the charge on the metal center and number of valence electrons • Group - Oxidation Number = d electron count 3 4 5 6 7 8 9 10 11 12 Size • Size of the metal ions follows periodic trends. • Higher positive charge generally means a smaller ion. • Higher on the periodic table means a smaller ion. • Ions of the same charge decrease in radius going across a row, e.g. Ca2+>Mn2+>Zn2+. • Of course, ion size will effect coordination number. 2 Coordination Number • Coordination Number = the number of donor atoms bound to the metal center. • The coordination number may or may not be equal to the number of ligands bound to the metal. • Different metal ions, depending on oxidation state prefer different coordination numbers. • Ligand size and electronic structure can also effect coordination number. Common Coordination Numbers • Low coordination numbers (n =2,3) are fairly rare, in biological systems a few examples are Cu(I) metallochaperones and Hg(II) metalloregulatory proteins. • Four-coordinate is fairly common in complexes of Cu(I/II), Zn(II), Co(II), as well as in biologically less relevant metal ions such as Pd(II) and Pt(II). • Five-coordinate is also fairly common, particularly for Fe(II). • Six-coordinate is the most common and important coordination number for most transition metal ions. • Higher coordination numbers are found in some 2nd and 3rd row transition metals, larger alkali metals, and lanthanides and actinides. -

Chapter 4, Bravais Lattice Primitive Vectors
Chapter 4, Bravais Lattice A Bravais lattice is the collection of all (and only those) points in space reachable from the origin with position vectors: n , n , n integer (+, -, or 0) r r r r 1 2 3 R = n1a1 + n2 a2 + n3a3 a1, a2, and a3 not all in same plane The three primitive vectors, a1, a2, and a3, uniquely define a Bravais lattice. However, for one Bravais lattice, there are many choices for the primitive vectors. A Bravais lattice is infinite. It is identical (in every aspect) when viewed from any of its lattice points. This is not a Bravais lattice. Honeycomb: P and Q are equivalent. R is not. A Bravais lattice can be defined as either the collection of lattice points, or the primitive translation vectors which construct the lattice. POINT Q OBJECT: Remember that a Bravais lattice has only points. Points, being dimensionless and isotropic, have full spatial symmetry (invariant under any point symmetry operation). Primitive Vectors There are many choices for the primitive vectors of a Bravais lattice. One sure way to find a set of primitive vectors (as described in Problem 4 .8) is the following: (1) a1 is the vector to a nearest neighbor lattice point. (2) a2 is the vector to a lattice points closest to, but not on, the a1 axis. (3) a3 is the vector to a lattice point nearest, but not on, the a18a2 plane. How does one prove that this is a set of primitive vectors? Hint: there should be no lattice points inside, or on the faces (lll)fhlhd(lllid)fd(parallolegrams) of, the polyhedron (parallelepiped) formed by these three vectors. -

Fifty Years of the VSEPR Model R.J
Available online at www.sciencedirect.com Coordination Chemistry Reviews 252 (2008) 1315–1327 Review Fifty years of the VSEPR model R.J. Gillespie Department of Chemistry, McMaster University, Hamilton, Ont. L8S 4M1, Canada Received 26 April 2007; accepted 21 July 2007 Available online 27 July 2007 Contents 1. Introduction ........................................................................................................... 1315 2. The VSEPR model ..................................................................................................... 1316 3. Force constants and the VSEPR model ................................................................................... 1318 4. Linnett’s double quartet model .......................................................................................... 1318 5. Exceptions to the VSEPR model, ligand–ligand repulsion and the ligand close packing (LCP) model ........................... 1318 6. Analysis of the electron density.......................................................................................... 1321 7. Molecules of the transition metals ....................................................................................... 1325 8. Teaching the VSEPR model ............................................................................................. 1326 9. Summary and conclusions .............................................................................................. 1326 Acknowledgements ................................................................................................... -

Multidisciplinary Design Project Engineering Dictionary Version 0.0.2
Multidisciplinary Design Project Engineering Dictionary Version 0.0.2 February 15, 2006 . DRAFT Cambridge-MIT Institute Multidisciplinary Design Project This Dictionary/Glossary of Engineering terms has been compiled to compliment the work developed as part of the Multi-disciplinary Design Project (MDP), which is a programme to develop teaching material and kits to aid the running of mechtronics projects in Universities and Schools. The project is being carried out with support from the Cambridge-MIT Institute undergraduate teaching programe. For more information about the project please visit the MDP website at http://www-mdp.eng.cam.ac.uk or contact Dr. Peter Long Prof. Alex Slocum Cambridge University Engineering Department Massachusetts Institute of Technology Trumpington Street, 77 Massachusetts Ave. Cambridge. Cambridge MA 02139-4307 CB2 1PZ. USA e-mail: [email protected] e-mail: [email protected] tel: +44 (0) 1223 332779 tel: +1 617 253 0012 For information about the CMI initiative please see Cambridge-MIT Institute website :- http://www.cambridge-mit.org CMI CMI, University of Cambridge Massachusetts Institute of Technology 10 Miller’s Yard, 77 Massachusetts Ave. Mill Lane, Cambridge MA 02139-4307 Cambridge. CB2 1RQ. USA tel: +44 (0) 1223 327207 tel. +1 617 253 7732 fax: +44 (0) 1223 765891 fax. +1 617 258 8539 . DRAFT 2 CMI-MDP Programme 1 Introduction This dictionary/glossary has not been developed as a definative work but as a useful reference book for engi- neering students to search when looking for the meaning of a word/phrase. It has been compiled from a number of existing glossaries together with a number of local additions. -

Searching Coordination Compounds
CAS ONLINEB Available on STN Internationalm The Scientific & Technical Information Network SEARCHING COORDINATION COMPOUNDS December 1986 Chemical Abstracts Service A Division of the American Chemical Society 2540 Olentangy River Road P.O. Box 3012 Columbus, OH 43210 Copyright O 1986 American Chemical Society Quoting or copying of material from this publication for educational purposes is encouraged. providing acknowledgment is made of the source of such material. SEARCHING COORDINATION COMPOUNDS prepared by Adrienne W. Kozlowski Professor of Chemistry Central Connecticut State University while on sabbatical leave as a Visiting Educator, Chemical Abstracts Service Table of Contents Topic PKEFACE ............................s.~........................ 1 CHAPTER 1: INTRODUCTION TO SEARCHING IN CAS ONLINE ............... 1 What is Substructure Searching? ............................... 1 The Basic Commands .............................................. 2 CHAPTEK 2: INTKOOUCTION TO COORDINATION COPPOUNDS ................ 5 Definitions and Terminology ..................................... 5 Ligand Characteristics.......................................... 6 Metal Characteristics .................................... ... 8 CHAPTEK 3: STKUCTUKING AND REGISTKATION POLICIES FOR COORDINATION COMPOUNDS .............................................11 Policies for Structuring Coordination Compounds ................. Ligands .................................................... Ligand Structures........................................... Metal-Ligand -

Symmetry and Groups, and Crystal Structures
CHAPTER 3: SYMMETRY AND GROUPS, AND CRYSTAL STRUCTURES Sarah Lambart RECAP CHAP. 2 2 different types of close packing: hcp: tetrahedral interstice (ABABA) ccp: octahedral interstice (ABCABC) Definitions: The coordination number or CN is the number of closest neighbors of opposite charge around an ion. It can range from 2 to 12 in ionic structures. These structures are called coordination polyhedron. RECAP CHAP. 2 Rx/Rz C.N. Type Hexagonal or An ideal close-packing of sphere 1.0 12 Cubic for a given CN, can only be Closest Packing achieved for a specific ratio of 1.0 - 0.732 8 Cubic ionic radii between the anions and 0.732 - 0.414 6 Octahedral Tetrahedral (ex.: the cations. 0.414 - 0.225 4 4- SiO4 ) 0.225 - 0.155 3 Triangular <0.155 2 Linear RECAP CHAP. 2 Pauling’s rule: #1: the coodination polyhedron is defined by the ratio Rcation/Ranion #2: The Electrostatic Valency (e.v.) Principle: ev = Z/CN #3: Shared edges and faces of coordination polyhedra decreases the stability of the crystal. #4: In crystal with different cations, those of high valency and small CN tend not to share polyhedral elements #5: The principle of parsimony: The number of different sites in a crystal tends to be small. CONTENT CHAP. 3 (2-3 LECTURES) Definitions: unit cell and lattice 7 Crystal systems 14 Bravais lattices Element of symmetry CRYSTAL LATTICE IN TWO DIMENSIONS A crystal consists of atoms, molecules, or ions in a pattern that repeats in three dimensions. The geometry of the repeating pattern of a crystal can be described in terms of a crystal lattice, constructed by connecting equivalent points throughout the crystal.