3885256.Pdf (2.220Mb)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Differential Expression Profiling of Gene Response to Ionizing Radiation in Two Endometrial Cancer Cell Lines with Distinct Radiosensitivities
625-634 28/1/2009 12:32 ÌÌ ™ÂÏ›‰·625 ONCOLOGY REPORTS 21: 625-634, 2009 625 Differential expression profiling of gene response to ionizing radiation in two endometrial cancer cell lines with distinct radiosensitivities XUE-LIAN DU1,2, TAO JIANG2, ZE-QING WEN1, QING-SHUI LI2, RONG GAO2 and FEI WANG1 1Department of Gynecologic Oncology, Shandong Tumor Hospital, Shandong University, Jinan 250117; 2Department of Obstetrics and Gynecology, Provincial Hospital Affiliated to Shandong University, Jinan 250021, P.R. China Received November 4, 2008; Accepted December 17, 2008 DOI: 10.3892/or_00000265 Abstracts. Although radiotherapy is routinely administered Introduction to high-risk endometrial carcinoma and offer a significant disease-free survival advantage, the therapeutic effect is Endometrial cancer is one of the most common gynecological sometimes limited by the occurrence of radioresistance. To malignancies worldwide. Surgery is the preferred initial determine the patterns of gene expression responsible for treatment and most women with early-stage, low-risk disease the radioresistance and to search for potential target genes for will do well without adjuvant radiotherapy. However, both radiotherapy, we selected two cell lines with distinct radio- intermediate-risk and high-risk patients are at risk for local- sensitivities using colony-formation assay from four endo- regional relapse and therefore adjuvant radiotherapy, such as metrial cancer cell lines. The cell cycle distribution showed pelvic radiation, vaginal brachytherapy, and whole-abdomen higher fractions of G2/M phase cells in the radiosensitive cell radiation, is essential for local control (1). Johnson and line KLE after radiation compared with the radioresistant cell colleagues reported that adjuvant external-beam pelvic radio- line ISK. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supporting Information
Supporting Information Edgar et al. 10.1073/pnas.1601895113 SI Methods (Actimetrics), and recordings were analyzed using LumiCycle Mice. Sample size was determined using the resource equation: Data Analysis software (Actimetrics). E (degrees of freedom in ANOVA) = (total number of exper- – Cell Cycle Analysis of Confluent Cell Monolayers. NIH 3T3, primary imental animals) (number of experimental groups), with −/− sample size adhering to the condition 10 < E < 20. For com- WT, and Bmal1 fibroblasts were sequentially transduced − − parison of MuHV-4 and HSV-1 infection in WT vs. Bmal1 / with lentiviral fluorescent ubiquitin-based cell cycle indicators mice at ZT7 (Fig. 2), the investigator did not know the genotype (FUCCI) mCherry::Cdt1 and amCyan::Geminin reporters (32). of the animals when conducting infections, bioluminescence Dual reporter-positive cells were selected by FACS (Influx Cell imaging, and quantification. For bioluminescence imaging, Sorter; BD Biosciences) and seeded onto 35-mm dishes for mice were injected intraperitoneally with endotoxin-free lucif- subsequent analysis. To confirm that expression of mCherry:: Cdt1 and amCyan::Geminin correspond to G1 (2n DNA con- erin (Promega E6552) using 2 mg total per mouse. Following < ≤ anesthesia with isofluorane, they were scanned with an IVIS tent) and S/G2 (2 n 4 DNA content) cell cycle phases, Lumina (Caliper Life Sciences), 15 min after luciferin admin- respectively, cells were stained with DNA dye DRAQ5 (abcam) and analyzed by flow cytometry (LSR-Fortessa; BD Biosci- istration. Signal intensity was quantified using Living Image ences). To examine dynamics of replicative activity under ex- software (Caliper Life Sciences), obtaining maximum radiance perimental confluent conditions, synchronized FUCCI reporter for designated regions of interest (photons per second per − − − monolayers were observed by time-lapse live cell imaging over square centimeter per Steradian: photons·s 1·cm 2·sr 1), relative 3 d (Nikon Eclipse Ti-E inverted epifluorescent microscope). -

Roles of TBC1D1 and TBC1D4 in Insulin- and Exercise-Stimulated Glucose Transport of Skeletal Muscle
Diabetologia (2015) 58:19–30 DOI 10.1007/s00125-014-3395-5 REVIEW Roles of TBC1D1 and TBC1D4 in insulin- and exercise-stimulated glucose transport of skeletal muscle Gregory D. Cartee Received: 30 June 2014 /Accepted: 7 August 2014 /Published online: 4 October 2014 # Springer-Verlag Berlin Heidelberg 2014 Abstract This review focuses on two paralogue Rab GTPase mechanism for greater TBC1D4 phosphorylation in insulin- activating proteins known as TBC1D1 Tre-2/BUB2/cdc 1 stimulated muscles after acute exercise is uncertain, and a domain family (TBC1D) 1 and TBC1D4 (also called Akt causal link between enhanced TBC1D4 phosphorylation and Substrate of 160 kDa, AS160) and their roles in controlling increased post-exercise insulin sensitivity has yet to be skeletal muscle glucose transport in response to the indepen- established. In summary, TBC1D1 and TBC1D4 have impor- dent and combined effects of insulin and exercise. Convincing tant, but distinct roles in regulating muscle glucose transport evidence implicates Akt2-dependent TBC1D4 phosphoryla- in response to insulin and exercise. tion on T642 as a key part of the mechanism for insulin- stimulated glucose uptake by skeletal muscle. TBC1D1 phos- Keywords Akt substrate of 160 kDa . Diabetes . Glucose phorylation on several insulin-responsive sites (including transport .High-fatdiet .Insulinresistance .Obesity .Physical T596, a site corresponding to T642 in TBC1D4) does not activity . Review appear to be essential for in vivo insulin-stimulated glucose uptake by skeletal muscle. In vivo exercise or ex vivo con- Abbreviations traction of muscle result in greater TBC1D1 phosphorylation AMPK 5' AMP-activated kinase on S237 that is likely to be secondary to increased AMP- APPL2 Adaptor protein containing PH domain, activated protein kinase activity and potentially important for PTB domain and leucine zipper motif 2 contraction-stimulated glucose uptake. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -
Download Validation Data
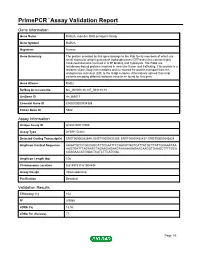
PrimePCR™Assay Validation Report Gene Information Gene Name RAB2A, member RAS oncogene family Gene Symbol RAB2A Organism Human Gene Summary The protein encoded by this gene belongs to the Rab family members of which are small molecular weight guanosine triphosphatases (GTPases) that contain highly conserved domains involved in GTP binding and hydrolysis. The Rabs are membrane-bound proteins involved in vesicular fusion and trafficking. This protein is a resident of pre-Golgi intermediates and is required for protein transport from the endoplasmic reticulum (ER) to the Golgi complex. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. Gene Aliases RAB2 RefSeq Accession No. NC_000008.10, NT_008183.19 UniGene ID Hs.369017 Ensembl Gene ID ENSG00000104388 Entrez Gene ID 5862 Assay Information Unique Assay ID qHsaCID0011980 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000262646, ENST00000531289, ENST00000452437, ENST00000543829 Amplicon Context Sequence AAGATGCCCGCCAGCATTCCAATTCCAACATGGTCATTATGCTTATTGGAAATAA AAGTGATTTAGAATCTAGAAGAGAAGTAAAAAAAGAAGAAGGTGAAGCTTTTGCA CGAGAACATGGACTCATCTTCATGGA Amplicon Length (bp) 106 Chromosome Location 8:61497317-61504494 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 104 R2 0.9988 cDNA Cq 18.53 cDNA Tm (Celsius) 77 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 43.2 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report -

A SARS-Cov-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug- Repurposing
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.22.002386; this version posted March 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug- Repurposing David E. Gordon1,2,3,4, Gwendolyn M. Jang1,2,3,4, Mehdi Bouhaddou1,2,3,4, Jiewei Xu1,2,3,4, Kirsten Obernier1,2,3,4, Matthew J. O'Meara5, Jeffrey Z. Guo1,2,3,4, Danielle L. Swaney1,2,3,4, Tia A. Tummino1,2,6, Ruth Huettenhain1,2,3,4, Robyn Kaake1,2,3,4, Alicia L. Richards1,2,3,4, Beril Tutuncuoglu1,2,3,4, Helene Foussard1,2,3,4, Jyoti Batra1,2,3,4, Kelsey Haas1,2,3,4, Maya Modak1,2,3,4, Minkyu Kim1,2,3,4, Paige Haas1,2,3,4, Benjamin J. Polacco1,2,3,4, Hannes Braberg1,2,3,4, Jacqueline M. Fabius1,2,3,4, Manon Eckhardt1,2,3,4, Margaret Soucheray1,2,3,4, Melanie J. Bennett1,2,3,4, Merve Cakir1,2,3,4, Michael J. McGregor1,2,3,4, Qiongyu Li1,2,3,4, Zun Zar Chi Naing1,2,3,4, Yuan Zhou1,2,3,4, Shiming Peng1,2,6, Ilsa T. Kirby1,4,7, James E. Melnyk1,4,7, John S. Chorba1,4,7, Kevin Lou1,4,7, ShiZhong A. Dai1,4,7, Wenqi Shen1,4,7, Ying Shi1,4,7, Ziyang Zhang1,4,7, Inigo Barrio-HernandeZ8, Danish Memon8, Claudia Hernandez-Armenta8, Christopher J.P. -
Primepcr™Assay Validation Report
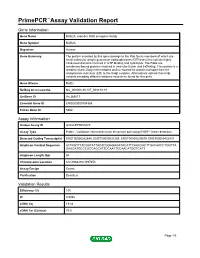
PrimePCR™Assay Validation Report Gene Information Gene Name RAB2A, member RAS oncogene family Gene Symbol RAB2A Organism Human Gene Summary The protein encoded by this gene belongs to the Rab family members of which are small molecular weight guanosine triphosphatases (GTPases) that contain highly conserved domains involved in GTP binding and hydrolysis. The Rabs are membrane-bound proteins involved in vesicular fusion and trafficking. This protein is a resident of pre-Golgi intermediates and is required for protein transport from the endoplasmic reticulum (ER) to the Golgi complex. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. Gene Aliases RAB2 RefSeq Accession No. NC_000008.10, NT_008183.19 UniGene ID Hs.369017 Ensembl Gene ID ENSG00000104388 Entrez Gene ID 5862 Assay Information Unique Assay ID qHsaCEP0050270 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000262646, ENST00000531289, ENST00000529579, ENST00000452437 Amplicon Context Sequence ACTAGTTTACGATATTACACGGAGAGATACATTCAACCACTTGACAACCTGGTTA GAAGATGCCCGCCAGCATTCCAATTCCAACATGGTCATT Amplicon Length (bp) 64 Chromosome Location 8:61496829-61497354 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 105 R2 0.9992 cDNA Cq 19.84 cDNA Tm (Celsius) 79.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 24.85 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay -

HOPS-Dependent Endosomal Fusion Required for Efficient Cytosolic Delivery of Therapeutic Peptides and Small Proteins
HOPS-dependent endosomal fusion required for efficient cytosolic delivery of therapeutic peptides and small proteins Angela Steinauera, Jonathan R. LaRochelleb, Susan L. Knoxa, Rebecca F. Wissnera, Samuel Berryc, and Alanna Schepartza,b,1 aDepartment of Chemistry, Yale University, New Haven, CT 06520-8107; bDepartment of Molecular, Cellular and Developmental Biology, Yale University, New Haven, CT 06520-8103; and cDepartment of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520-8114 Edited by James A. Wells, University of California, San Francisco, CA, and approved November 26, 2018 (received for review July 17, 2018) Protein therapeutics represent a significant and growing compo- Recently, we discovered that, when added to cells, certain small, nent of the modern pharmacopeia, but their potential to treat folded miniature proteins (9, 10) derived from avian pancreatic human disease is limited because most proteins fail to traffic polypeptide (aPP) or an isolated zinc-finger (ZF) domain, are across biological membranes. Recently, we discovered a class of taken up into the endocytic pathway and subsequently released cell-permeant miniature proteins (CPMPs) containing a precisely into the cytosol with unprecedented efficiencies (11, 12). The most defined, penta-arginine (penta-Arg) motif that traffics readily to effective molecules are defined by a discrete array of five arginine the cytosol and nucleus of mammalian cells with efficiencies that residues on a folded α-helix (13); we refer to these molecules as rival those of hydrocarbon-stapled peptides active in animals and cell-permeant miniature proteins (CPMPs). Treatment of HeLa man. Like many cell-penetrating peptides (CPPs), CPMPs enter the cells in culture with the CPMP ZF5.3 leads to a ZF5.3 concen- endocytic pathway; the difference is that CPMPs containing a penta- tration in the cytosol that is roughly 67% of the extracellular in- Arg motif are released efficiently from endosomes, while other CPPs cubation concentration; this value is at least 10-fold higher than are not. -

A SARS-Cov-2 Protein Interaction Map Reveals Targets for Drug Repurposing
Article A SARS-CoV-2 protein interaction map reveals targets for drug repurposing https://doi.org/10.1038/s41586-020-2286-9 A list of authors and affiliations appears at the end of the paper Received: 23 March 2020 Accepted: 22 April 2020 A newly described coronavirus named severe acute respiratory syndrome Published online: 30 April 2020 coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than Check for updates 160,000 individuals and caused worldwide social and economic disruption1,2. There are no antiviral drugs with proven clinical efcacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and eforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identifed the human proteins that physically associated with each of the SARS-CoV-2 proteins using afnity-purifcation mass spectrometry, identifying 332 high-confdence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors. -
![Rab2 Antibody [Discontinued, View Alternatives] (R31430)](https://docslib.b-cdn.net/cover/7281/rab2-antibody-discontinued-view-alternatives-r31430-2777281.webp)
Rab2 Antibody [Discontinued, View Alternatives] (R31430)
Rab2 Antibody [Discontinued, view alternatives] (R31430) Catalog No. Formulation Size R31430 0.5mg/ml if reconstituted with 0.2ml sterile DI water 100 ug Bulk quote request Availability Discontinued Species Reactivity Human, Mouse, Rat Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit IgG Purity Antigen affinity Buffer Lyophilized from 1X PBS with 2.5% BSA and 0.025% sodium azide/thimerosal UniProt P61019 Applications Western blot : 0.5-1ug/ml Limitations This Rab2 antibody is available for research use only. Western blot testing of Rab2 antibody and Lane 1: rat brain; 2: PC12 lysate Description Ras-related protein Rab-2A is a protein that in humans is encoded by the RAB2A gene. The protein encoded by this gene belongs to the Rab family, members of which are small molecular weight guanosine triphosphatases(GTPases) that contain highly conserved domains involved in GTP binding and hydrolysis. It is mapped to 8q12.1. RAB2A protein is a resident of pre-Golgi intermediates and is required for protein transport from the endoplasmic reticulum to the Golgi complex. It is essential for the maturation of pre-Golgi intermediates. RAB2A has been shown to interact with GOLGA2. Application Notes The stated application concentrations are suggested starting amounts. Titration of the Rab2 antibody may be required due to differences in protocols and secondary/substrate sensitivity. Immunogen An amino acid sequence from the C-terminus of human RAB2A (QHAATNATHAGNQGGQQA) was used as the immunogen for this Rab2 antibody. Storage After reconstitution, the Rab2 antibody can be stored for up to one month at 4oC. For long-term, aliquot and store at -20oC. -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV).