The Boiling Springs Lake Metavirome: Charting the Viral Sequence-Space of an Extreme Environment Microbial Ecosystem
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Viral Haemorrhagic Septicaemia Virus (VHSV): on the Search for Determinants Important for Virulence in Rainbow Trout Oncorhynchus Mykiss
Downloaded from orbit.dtu.dk on: Nov 08, 2017 Viral haemorrhagic septicaemia virus (VHSV): on the search for determinants important for virulence in rainbow trout oncorhynchus mykiss Olesen, Niels Jørgen; Skall, H. F.; Kurita, J.; Mori, K.; Ito, T. Published in: 17th International Conference on Diseases of Fish And Shellfish Publication date: 2015 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Olesen, N. J., Skall, H. F., Kurita, J., Mori, K., & Ito, T. (2015). Viral haemorrhagic septicaemia virus (VHSV): on the search for determinants important for virulence in rainbow trout oncorhynchus mykiss. In 17th International Conference on Diseases of Fish And Shellfish: Abstract book (pp. 147-147). [O-139] Las Palmas: European Association of Fish Pathologists. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. DISCLAIMER: The organizer takes no responsibility for any of the content stated in the abstracts. -

Oryzias Latipes)
Betanodavirus infection in the freshwater model fish medaka (Oryzias latipes) Ryo Furusawa, Yasushi Okinaka,* and Toshihiro Nakai Graduate School of Biosphere Science, Hiroshima University, Higashi-hiroshima 739- 8528, Japan æfAuthor for correspondence: Yasushi Okinaka. Telephone: +8 1-82-424-7978. Fax: +81- 82-424-79 1 6. E-mail: [email protected] Running title: Betanodavirus infection in medaka Key words: medaka, betanodavirus, model fish, model virus, freshwater fish Total number of words; text (3532 words), summary (230 words) Total number of figures; 6 figures Total number of tables; 0 table SUMMARY Betanodaviruses, the causal agents of viral nervous necrosis in marine fish, have bipartite positive-sense RNA genomes. Because the genomes are the smallest and simplest among viruses, betanodaviruses are well studied using a genetic engineering system as model viruses, like the cases with the insect viruses, alphanodaviruses, the other members of the family Nodaviridae. However, studies of virus-host interactions have been limited because betanodaviruses basically infect marine fish at early developmental stages (larval and juvenile). These fish are only available for a few months of the year and are not suitable for the construction of a reversed genetics system. To overcome these problems, several freshwater fish species were tested for their susceptibility to betanodaviruses. We have demonstrated that adult medaka (Oryzias latipes), a well-known model fish, is susceptible to both Stripedjack nervous necrosis virus (the type species of the betanodaviruses) and Redspotted grouper nervous necrosis virus which have different host specificity in marine fish species. Infected medaka exhibited erratic swimming and the viruses were specifically localized to the brain, spinal cord, and retina of the infected fish, similar to the pattern of infection in naturally infected marine fish. -

Evidence for Viral Infection in the Copepods Labidocera Aestiva And
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2012 Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida Darren Stephenson Dunlap University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the American Studies Commons, Other Oceanography and Atmospheric Sciences and Meteorology Commons, and the Virology Commons Scholar Commons Citation Dunlap, Darren Stephenson, "Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida" (2012). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4032 This Thesis is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Evidence of Viruses in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida By Darren S. Dunlap A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science College of Marine Science University of South Florida Major Professor: Mya Breitbart, Ph.D Kendra Daly, Ph.D Ian Hewson, Ph.D Date of Approval: March 19, 2012 Key Words: Copepods, Single-stranded DNA Viruses, Mesozooplankton, Transmission Electron Microscopy, Metagenomics Copyright © 2012, Darren Stephenson Dunlap DEDICATION None of this would have been possible without the generous love and support of my entire family over the years. My parents, Steve and Jill Dunlap, have always encouraged my pursuits with support and love, and their persistence of throwing me into lakes and rivers is largely responsible for my passion for Marine Science. -

Frequent Occurrence of Mungbean Yellow Mosaic India Virus in Tomato Leaf Curl Disease Afected Tomato in Oman M
www.nature.com/scientificreports OPEN Frequent occurrence of Mungbean yellow mosaic India virus in tomato leaf curl disease afected tomato in Oman M. S. Shahid 1*, M. Shafq 1, M. Ilyas2, A. Raza1, M. N. Al-Sadrani1, A. M. Al-Sadi 1 & R. W. Briddon 3 Next generation sequencing (NGS) of DNAs amplifed by rolling circle amplifcation from 6 tomato (Solanum lycopersicum) plants with leaf curl symptoms identifed a number of monopartite begomoviruses, including Tomato yellow leaf curl virus (TYLCV), and a betasatellite (Tomato leaf curl betasatellite [ToLCB]). Both TYLCV and ToLCB have previously been identifed infecting tomato in Oman. Surprisingly the NGS results also suggested the presence of the bipartite, legume-adapted begomovirus Mungbean yellow mosaic Indian virus (MYMIV). The presence of MYMIV was confrmed by cloning and Sanger sequencing from four of the six plants. A wider analysis by PCR showed MYMIV infection of tomato in Oman to be widespread. Inoculation of plants with full-length clones showed the host range of MYMIV not to extend to Nicotiana benthamiana or tomato. Inoculation to N. benthamiana showed TYLCV to be capable of maintaining MYMIV in both the presence and absence of the betasatellite. In tomato MYMIV was only maintained by TYLCV in the presence of the betasatellite and then only at low titre and efciency. This is the frst identifcation of TYLCV with ToLCB and the legume adapted bipartite begomovirus MYMIV co-infecting tomato. This fnding has far reaching implications. TYLCV has spread around the World from its origins in the Mediterranean/Middle East, in some instances, in live tomato planting material. -

Emerging Viral Diseases of Fish and Shrimp Peter J
Emerging viral diseases of fish and shrimp Peter J. Walker, James R. Winton To cite this version: Peter J. Walker, James R. Winton. Emerging viral diseases of fish and shrimp. Veterinary Research, BioMed Central, 2010, 41 (6), 10.1051/vetres/2010022. hal-00903183 HAL Id: hal-00903183 https://hal.archives-ouvertes.fr/hal-00903183 Submitted on 1 Jan 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Vet. Res. (2010) 41:51 www.vetres.org DOI: 10.1051/vetres/2010022 Ó INRA, EDP Sciences, 2010 Review article Emerging viral diseases of fish and shrimp 1 2 Peter J. WALKER *, James R. WINTON 1 CSIRO Livestock Industries, Australian Animal Health Laboratory (AAHL), 5 Portarlington Road, Geelong, Victoria, Australia 2 USGS Western Fisheries Research Center, 6505 NE 65th Street, Seattle, Washington, USA (Received 7 December 2009; accepted 19 April 2010) Abstract – The rise of aquaculture has been one of the most profound changes in global food production of the past 100 years. Driven by population growth, rising demand for seafood and a levelling of production from capture fisheries, the practice of farming aquatic animals has expanded rapidly to become a major global industry. -
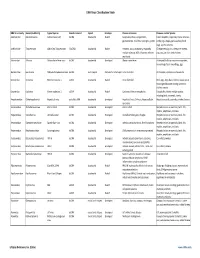
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae -

Viruses in Transplantation - Not Always Enemies
Viruses in transplantation - not always enemies Virome and transplantation ECCMID 2018 - Madrid Prof. Laurent Kaiser Head Division of Infectious Diseases Laboratory of Virology Geneva Center for Emerging Viral Diseases University Hospital of Geneva ESCMID eLibrary © by author Conflict of interest None ESCMID eLibrary © by author The human virome: definition? Repertoire of viruses found on the surface of/inside any body fluid/tissue • Eukaryotic DNA and RNA viruses • Prokaryotic DNA and RNA viruses (phages) 25 • The “main” viral community (up to 10 bacteriophages in humans) Haynes M. 2011, Metagenomic of the human body • Endogenous viral elements integrated into host chromosomes (8% of the human genome) • NGS is shaping the definition Rascovan N et al. Annu Rev Microbiol 2016;70:125-41 Popgeorgiev N et al. Intervirology 2013;56:395-412 Norman JM et al. Cell 2015;160:447-60 ESCMID eLibraryFoxman EF et al. Nat Rev Microbiol 2011;9:254-64 © by author Viruses routinely known to cause diseases (non exhaustive) Upper resp./oropharyngeal HSV 1 Influenza CNS Mumps virus Rhinovirus JC virus RSV Eye Herpes viruses Parainfluenza HSV Measles Coronavirus Adenovirus LCM virus Cytomegalovirus Flaviviruses Rabies HHV6 Poliovirus Heart Lower respiratory HTLV-1 Coxsackie B virus Rhinoviruses Parainfluenza virus HIV Coronaviruses Respiratory syncytial virus Parainfluenza virus Adenovirus Respiratory syncytial virus Coronaviruses Gastro-intestinal Influenza virus type A and B Human Bocavirus 1 Adenovirus Hepatitis virus type A, B, C, D, E Those that cause -

Viral Nanoparticles and Virus-Like Particles: Platforms for Contemporary Vaccine Design Emily M
Advanced Review Viral nanoparticles and virus-like particles: platforms for contemporary vaccine design Emily M. Plummer1,2 and Marianne Manchester2∗ Current vaccines that provide protection against infectious diseases have primarily relied on attenuated or inactivated pathogens. Virus-like particles (VLPs), comprised of capsid proteins that can initiate an immune response but do not include the genetic material required for replication, promote immunogenicity and have been developed and approved as vaccines in some cases. In addition, many of these VLPs can be used as molecular platforms for genetic fusion or chemical attachment of heterologous antigenic epitopes. This approach has been shown to provide protective immunity against the foreign epitopes in many cases. A variety of VLPs and virus-based nanoparticles are being developed for use as vaccines and epitope platforms. These particles have the potential to increase efficacy of current vaccines as well as treat diseases for which no effective vaccines are available. 2010 John Wiley & Sons, Inc. WIREs Nanomed Nanobiotechnol 2011 3 174–196 DOI: 10.1002/wnan.119 INTRODUCTION are normally associated with virus infection. PAMPs can be recognized by Toll-like receptors (TLRs) and he goal of vaccination is to initiate a strong other pattern-recognition receptors (PRRs) which Timmune response that leads to the development are present on the surface of host cells.4 The of lasting and protective immunity. Vaccines against intrinsic properties of multivalent display and highly pathogens are the most common, but approaches to ordered structure present in many pathogens also develop vaccines against cancer cells, host proteins, or 1,2 facilitate recognition by PAMPs, resulting in increased small molecule drugs have been developed as well. -

Identification of a Nanovirus-Alphasatellite Complex in Sophora Alopecuroides
Accepted Manuscript Title: Identification of a nanovirus-alphasatellite complex in Sophora alopecuroides Author: Jahangir Heydarnejad Mehdi Kamali Hossain Massumi Anders Kvarnheden Maketalena F. Male Simona Kraberger Daisy Stainton Darren P. Martin Arvind Varsani PII: S0168-1702(17)30009-6 DOI: http://dx.doi.org/doi:10.1016/j.virusres.2017.03.023 Reference: VIRUS 97113 To appear in: Virus Research Received date: 5-1-2017 Revised date: 15-3-2017 Accepted date: 18-3-2017 Please cite this article as: Heydarnejad, J., Kamali, M., Massumi, H., Kvarnheden, A., Male, M.F., Kraberger, S., Stainton, D., Martin, D.P., Varsani, A.,Identification of a nanovirus-alphasatellite complex in Sophora alopecuroides, Virus Research (2017), http://dx.doi.org/10.1016/j.virusres.2017.03.023 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. 1 Identification of a nanovirus-alphasatellite complex in Sophora alopecuroides 2 Jahangir Heydarnejad 1* , Mehdi Kamali 1, Hossain Massumi 1, Anders Kvarnheden 2, Maketalena 3 F. Male 3, Simona Kraberger 3,4 , Daisy Stainton 3,5 , Darren P. Martin 6 and Arvind Varsani 3,7,8* 4 5 1Department of Plant Protection, College -

Viruses Virus Diseases Poaceae(Gramineae)
Viruses and virus diseases of Poaceae (Gramineae) Viruses The Poaceae are one of the most important plant families in terms of the number of species, worldwide distribution, ecosystems and as ingredients of human and animal food. It is not surprising that they support many parasites including and more than 100 severely pathogenic virus species, of which new ones are being virus diseases regularly described. This book results from the contributions of 150 well-known specialists and presents of for the first time an in-depth look at all the viruses (including the retrotransposons) Poaceae(Gramineae) infesting one plant family. Ta xonomic and agronomic descriptions of the Poaceae are presented, followed by data on molecular and biological characteristics of the viruses and descriptions up to species level. Virus diseases of field grasses (barley, maize, rice, rye, sorghum, sugarcane, triticale and wheats), forage, ornamental, aromatic, wild and lawn Gramineae are largely described and illustrated (32 colour plates). A detailed index Sciences de la vie e) of viruses and taxonomic lists will help readers in their search for information. Foreworded by Marc Van Regenmortel, this book is essential for anyone with an interest in plant pathology especially plant virology, entomology, breeding minea and forecasting. Agronomists will also find this book invaluable. ra The book was coordinated by Hervé Lapierre, previously a researcher at the Institut H. Lapierre, P.-A. Signoret, editors National de la Recherche Agronomique (Versailles-France) and Pierre A. Signoret emeritus eae (G professor and formerly head of the plant pathology department at Ecole Nationale Supérieure ac Agronomique (Montpellier-France). Both have worked from the late 1960’s on virus diseases Po of Poaceae . -

Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics
viruses Article Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics Ashleigh F. Porter 1, Mang Shi 1, John-Sebastian Eden 1,2 , Yong-Zhen Zhang 3,4 and Edward C. Holmes 1,3,* 1 Marie Bashir Institute for Infectious Diseases and Biosecurity, Charles Perkins Centre, School of Life & Environmental Sciences and Sydney Medical School, The University of Sydney, Sydney, NSW 2006, Australia; [email protected] (A.F.P.); [email protected] (M.S.); [email protected] (J.-S.E.) 2 Centre for Virus Research, Westmead Institute for Medical Research, Westmead, NSW 2145, Australia 3 Shanghai Public Health Clinical Center and School of Public Health, Fudan University, Shanghai 201500, China; [email protected] 4 Department of Zoonosis, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Changping, Beijing 102206, China * Correspondence: [email protected]; Tel.: +61-2-9351-5591 Received: 17 October 2019; Accepted: 23 November 2019; Published: 25 November 2019 Abstract: DNA viruses comprise a wide array of genome structures and infect diverse host species. To date, most studies of DNA viruses have focused on those with the strongest disease associations. Accordingly, there has been a marked lack of sampling of DNA viruses from invertebrates. Bulk RNA sequencing has resulted in the discovery of a myriad of novel RNA viruses, and herein we used this methodology to identify actively transcribing DNA viruses in meta-transcriptomic libraries of diverse invertebrate species. Our analysis revealed high levels of phylogenetic diversity in DNA viruses, including 13 species from the Parvoviridae, Circoviridae, and Genomoviridae families of single-stranded DNA virus families, and six double-stranded DNA virus species from the Nudiviridae, Polyomaviridae, and Herpesviridae, for which few invertebrate viruses have been identified to date. -

An Unusual Alphasatellite Associated with Monopartite Begomoviruses Attenuates Symptoms and Reduces Betasatellite Accumulation
Journal of General Virology (2011), 92, 706–717 DOI 10.1099/vir.0.025288-0 An unusual alphasatellite associated with monopartite begomoviruses attenuates symptoms and reduces betasatellite accumulation Ali M. Idris,1,2 M. Shafiq Shahid,1,3 Rob W. Briddon,3 A. J. Khan,4 J.-K. Zhu2 and J. K. Brown1 Correspondence 1School of Plant Sciences, The University of Arizona, Tucson, AZ 85721, USA J. K. Brown 2Plant Stress Genomics Research Center, King Abdullah University of Science and Technology, [email protected] Thuwal 23955-6900, Kingdom of Saudi Arabia 3National Institute for Biotechnology and Genetic Engineering, PO Box 577, Jhang Road, Faisalabad, Pakistan 4Department of Crop Sciences, College of Agricultural and Marine Sciences, Sultan Qaboos University, PO Box 34, Al-Khod 123, Muscat, Sultanate of Oman The Oman strain of Tomato yellow leaf curl virus (TYLCV-OM) and its associated betasatellite, an isolate of Tomato leaf curl betasatellite (ToLCB), were previously reported from Oman. Here we report the isolation of a second, previously undescribed, begomovirus [Tomato leaf curl Oman virus (ToLCOMV)] and an alphasatellite from that same plant sample. This alphasatellite is closely related (90 % shared nucleotide identity) to an unusual DNA-2-type Ageratum yellow vein Singapore alphasatellite (AYVSGA), thus far identified only in Singapore. ToLCOMV was found to have a recombinant genome comprising sequences derived from two extant parents, TYLCV-OM, which is indigenous to Oman, and Papaya leaf curl virus from the Indian subcontinent. All possible combinations of ToLCOMV, TYLCV-OM, ToLCB and AYVSGA were used to agro-inoculate tomato and Nicotiana benthamiana. Infection with ToLCOMV yielded mild leaf-curl symptoms in both hosts; however, plants inoculated with TYLCV-OM developed more severe symptoms.