Single Stranded DNA Viruses Associated with Capybara Faeces Sampled in Brazil
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Elisabeth Mendes Martins De Moura Diversidade De Vírus DNA
Elisabeth Mendes Martins de Moura Diversidade de vírus DNA autóctones e alóctones de mananciais e de esgoto da região metropolitana de São Paulo Tese apresentada ao Programa de Pós- Graduação em Microbiologia do Instituto de Ciências Biomédicas da Universidade de São Paulo, para obtenção do Titulo de Doutor em Ciências. Área de concentração: Microbiologia Orienta: Prof (a). Dr (a). Dolores Ursula Mehnert versão original São Paulo 2017 RESUMO MOURA, E. M. M. Diversidade de vírus DNA autóctones e alóctones de mananciais e de esgoto da região metropolitana de São Paulo. 2017. 134f. Tese (Doutorado em Microbiologia) - Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, 2017. A água doce no Brasil, assim como o seu consumo é extremamente importante para as diversas atividades criadas pelo ser humano. Por esta razão o consumo deste bem é muito grande e consequentemente, provocando o seu impacto. Os mananciais são normalmente usados para abastecimento doméstico, comercial, industrial e outros fins. Os estudos na área de ecologia de micro-organismos nos ecossistemas aquáticos (mananciais) e em esgotos vêm sendo realizados com mais intensidade nos últimos anos. Nas últimas décadas foi introduzido o conceito de virioplâncton com base na abundância e diversidade de partículas virais presentes no ambiente aquático. O virioplâncton influencia muitos processos ecológicos e biogeoquímicos, como ciclagem de nutriente, taxa de sedimentação de partículas, diversidade e distribuição de espécies de algas e bactérias, controle de florações de fitoplâncton e transferência genética horizontal. Os estudos nesta área da virologia molecular ainda estão muito restritos no país, bem como muito pouco se conhece sobre a diversidade viral na água no Brasil. -

Evidence for Viral Infection in the Copepods Labidocera Aestiva And
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2012 Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida Darren Stephenson Dunlap University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the American Studies Commons, Other Oceanography and Atmospheric Sciences and Meteorology Commons, and the Virology Commons Scholar Commons Citation Dunlap, Darren Stephenson, "Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida" (2012). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4032 This Thesis is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Evidence of Viruses in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida By Darren S. Dunlap A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science College of Marine Science University of South Florida Major Professor: Mya Breitbart, Ph.D Kendra Daly, Ph.D Ian Hewson, Ph.D Date of Approval: March 19, 2012 Key Words: Copepods, Single-stranded DNA Viruses, Mesozooplankton, Transmission Electron Microscopy, Metagenomics Copyright © 2012, Darren Stephenson Dunlap DEDICATION None of this would have been possible without the generous love and support of my entire family over the years. My parents, Steve and Jill Dunlap, have always encouraged my pursuits with support and love, and their persistence of throwing me into lakes and rivers is largely responsible for my passion for Marine Science. -

Pdf Available
Virology 554 (2021) 89–96 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/virology Diverse cressdnaviruses and an anellovirus identifiedin the fecal samples of yellow-bellied marmots Anthony Khalifeh a, Daniel T. Blumstein b,**, Rafaela S. Fontenele a, Kara Schmidlin a, C´ecile Richet a, Simona Kraberger a, Arvind Varsani a,c,* a The Biodesign Center for Fundamental and Applied Microbiomics, School of Life Sciences, Center for Evolution and Medicine, Arizona State University, Tempe, AZ, 85287, USA b Department of Ecology & Evolutionary Biology, Institute of the Environment & Sustainability, University of California Los Angeles, Los Angeles, CA, 90095, USA c Structural Biology Research Unit, Department of Clinical Laboratory Sciences, University of Cape Town, 7925, Cape Town, South Africa ARTICLE INFO ABSTRACT Keywords: Over that last decade, coupling multiple strand displacement approaches with high throughput sequencing have Marmota flaviventer resulted in the identification of genomes of diverse groups of small circular DNA viruses. Using a similar Anelloviridae approach but with recovery of complete genomes by PCR, we identified a diverse group of single-stranded vi Genomoviridae ruses in yellow-bellied marmot (Marmota flaviventer) fecal samples. From 13 fecal samples we identified viruses Cressdnaviricota in the family Genomoviridae (n = 7) and Anelloviridae (n = 1), and several others that ware part of the larger Cressdnaviricota phylum but not within established families (n = 19). There were also circular DNA molecules identified (n = 4) that appear to encode one viral-like gene and have genomes of <1545 nts. This study gives a snapshot of viruses associated with marmots based on fecal sampling. -

Viral Diversity in Oral Cavity from Sapajus Nigritus by Metagenomic Analyses
Brazilian Journal of Microbiology (2020) 51:1941–1951 https://doi.org/10.1007/s42770-020-00350-w ENVIRONMENTAL MICROBIOLOGY - RESEARCH PAPER Viral diversity in oral cavity from Sapajus nigritus by metagenomic analyses Raissa Nunes dos Santos1,2 & Fabricio Souza Campos2,3 & Fernando Finoketti1,2 & Anne Caroline dos Santos1,2 & Aline Alves Scarpellini Campos1,2,3 & Paulo Guilherme Carniel Wagner2,4 & Paulo Michel Roehe 1,2 & Helena Beatriz de Carvalho Ruthner Batista2,5 & Ana Claudia Franco1,2 Received: 20 January 2020 /Accepted: 25 July 2020 / Published online: 11 August 2020 # Sociedade Brasileira de Microbiologia 2020 Abstract Sapajus nigritus are non-human primates which are widespread in South America. They are omnivores and live in troops of up to 40 individuals. The oral cavity is one of the main entry routes for microorganisms, including viruses. Our study proposed the identification of viral sequences from oral swabs collected in a group of capuchin monkeys (n = 5) living in a public park in a fragment of Mata Atlantica in South Brazil. Samples were submitted to nucleic acid extraction and enrichment, which was followed by the construction of libraries. After high-throughput sequencing and contig assembly, we used a pipeline to identify 11 viral families, which are Herpesviridae, Parvoviridae, Papillomaviridae, Polyomaviridae, Caulimoviridae, Iridoviridae, Astroviridae, Poxviridae,andBaculoviridae, in addition to two complete viral genomes of Anelloviridae and Genomoviridae. Some of these viruses were closely related to known viruses, while other fragments are more distantly related, with 50% of identity or less to the currently available virus sequences in databases. In addition to host-related viruses, insect and small vertebrate-related viruses were also found, as well as plant-related viruses, bringing insights about their diet. -

Multiple Origins of Prokaryotic and Eukaryotic Single-Stranded DNA Viruses from Bacterial and Archaeal Plasmids
ARTICLE https://doi.org/10.1038/s41467-019-11433-0 OPEN Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids Darius Kazlauskas 1, Arvind Varsani 2,3, Eugene V. Koonin 4 & Mart Krupovic 5 Single-stranded (ss) DNA viruses are a major component of the earth virome. In particular, the circular, Rep-encoding ssDNA (CRESS-DNA) viruses show high diversity and abundance 1234567890():,; in various habitats. By combining sequence similarity network and phylogenetic analyses of the replication proteins (Rep) belonging to the HUH endonuclease superfamily, we show that the replication machinery of the CRESS-DNA viruses evolved, on three independent occa- sions, from the Reps of bacterial rolling circle-replicating plasmids. The CRESS-DNA viruses emerged via recombination between such plasmids and cDNA copies of capsid genes of eukaryotic positive-sense RNA viruses. Similarly, the rep genes of prokaryotic DNA viruses appear to have evolved from HUH endonuclease genes of various bacterial and archaeal plasmids. Our findings also suggest that eukaryotic polyomaviruses and papillomaviruses with dsDNA genomes have evolved via parvoviruses from CRESS-DNA viruses. Collectively, our results shed light on the complex evolutionary history of a major class of viruses revealing its polyphyletic origins. 1 Institute of Biotechnology, Life Sciences Center, Vilnius University, Saulėtekio av. 7, Vilnius 10257, Lithuania. 2 The Biodesign Center for Fundamental and Applied Microbiomics, School of Life Sciences, Center for Evolution and Medicine, Arizona State University, Tempe, AZ 85287, USA. 3 Structural Biology Research Unit, Department of Integrative Biomedical Sciences, University of Cape Town, Rondebosch, 7700 Cape Town, South Africa. -

Small Circular DNA Viruses: the Muddy Viral “Playground” of Recombinant, Reassortant, and Highly Diverse Viruses
Small Circular DNA Viruses: The muddy viral “playground” of recombinant, reassortant, and highly diverse viruses @Varsani_lab Arizona State University, USA University of Canterbury, New Zealand University of Cape Town, South Africa Arvind Varsani Nucleotide substitutions Recombination Reassortment Viral evolution Viral Nucleotide substitutions Recombination Reassortment Viral evolution Viral Nucleotide substitutions Recombination Reassortment Viral evolution Viral I II III IV V dsDNA ssDNA VI VII dsRNA ssRNA(+) ssRNA(-) ssRNA(RT) dsDNA(RT) viruses Archaea Bacteria Diatoms Fungi Invertebrates Plants Vertebrates Unknown Anelloviridae – circular (~2.1 - 3.8kb) Bacillidnaviridae - circular (~4.5 - 6kb) Circoviridae – circular (~1.6 - 2.3kb) Geminiviridae – circular (~2.7kb; monopartite, bipartite, associated with α/β satellites) Genomovirdae – circular (1.8 -2.3 kb) ssDNA Inoviridae – circular (~5.8 - 8.7kb) Microviridae – circular (~4.5 - 6kb) Nanoviridae – circular (~1.2kb; multicomponent viruses; associated with satellites) Parvoviridae – linear (~3.7 - 6kb) Pleolipoviridae* – circular ssDNA; circular dsDNA or liner dsDNA (7-16kb) Smacoviridae* - circular (2.3 - 2.9kb) ssDNA viruses Chimerism in Rep Geminiviruses MSV: ~2 x 10-4 subs/site/year SSRV: ~3 x 10-4 subs/site/year EACMV: ~1 x 10-3 subs/site/year TYLCV: ~3 x 10-4 subs/site/year Nanoviruses FBNYV: ~1 x 10-3 subs/site/year BBTV: ~3 x 10-4 subs/site/year Circoviruses BFDV: ~2 x 10-3 subs/site/year PCV-2: ~1 x 10-3 subs/site/year Parvoviruses CPV: ~2 x 10-4 - 8 x 10-5 subs/site/year -

The LUCA and Its Complex Virome in Another Recent Synthesis, We Examined the Origins of the Replication and Structural Mart Krupovic , Valerian V
PERSPECTIVES archaea that form several distinct, seemingly unrelated groups16–18. The LUCA and its complex virome In another recent synthesis, we examined the origins of the replication and structural Mart Krupovic , Valerian V. Dolja and Eugene V. Koonin modules of viruses and posited a ‘chimeric’ scenario of virus evolution19. Under this Abstract | The last universal cellular ancestor (LUCA) is the most recent population model, the replication machineries of each of of organisms from which all cellular life on Earth descends. The reconstruction of the four realms derive from the primordial the genome and phenotype of the LUCA is a major challenge in evolutionary pool of genetic elements, whereas the major biology. Given that all life forms are associated with viruses and/or other mobile virion structural proteins were acquired genetic elements, there is no doubt that the LUCA was a host to viruses. Here, by from cellular hosts at different stages of evolution giving rise to bona fide viruses. projecting back in time using the extant distribution of viruses across the two In this Perspective article, we combine primary domains of life, bacteria and archaea, and tracing the evolutionary this recent work with observations on the histories of some key virus genes, we attempt a reconstruction of the LUCA virome. host ranges of viruses in each of the four Even a conservative version of this reconstruction suggests a remarkably complex realms, along with deeper reconstructions virome that already included the main groups of extant viruses of bacteria and of virus evolution, to tentatively infer archaea. We further present evidence of extensive virus evolution antedating the the composition of the virome of the last universal cellular ancestor (LUCA; also LUCA. -

Virus–Host Interactions and Their Roles in Coral Reef Health and Disease
!"#$"%& Virus–host interactions and their roles in coral reef health and disease Rebecca Vega Thurber1, Jérôme P. Payet1,2, Andrew R. Thurber1,2 and Adrienne M. S. Correa3 !"#$%&'$()(*+%&,(%--.#(+''/%!01(1/$%0-1$23++%(#4&,,+5(5&$-%#6('+1#$0$/$-("0+708-%#0$9(&17( 3%+7/'$080$9(4+$#3+$#6(&17(&%-($4%-&$-1-7("9(&1$4%+3+:-10'(70#$/%"&1'-;(<40#(=-80-5(3%+807-#( &1(01$%+7/'$0+1($+('+%&,(%--.(80%+,+:9(&17(->34�?-#($4-(,01@#("-$5--1(80%/#-#6('+%&,(>+%$&,0$9( &17(%--.(-'+#9#$->(7-',01-;(A-(7-#'%0"-($4-(70#$01'$08-("-1$40'2&##+'0&$-7(&17(5&$-%2'+,/>12( &##+'0&$-7(80%+>-#($4&$(&%-(/10B/-($+('+%&,(%--.#6(540'4(4&8-(%-'-08-7(,-##(&$$-1$0+1($4&1( 80%/#-#(01(+3-12+'-&1(#9#$->#;(A-(493+$4-#0?-($4&$(80%/#-#(+.("&'$-%0&(&17(-/@&%9+$-#( 791&>0'&,,9(01$-%&'$(50$4($4-0%(4+#$#(01($4-(5&$-%('+,/>1(&17(50$4(#',-%&'$010&1(C#$+19D('+%&,#($+( 01.,/-1'-(>0'%+"0&,('+>>/10$9(791&>0'#6('+%&,(",-&'401:(&17(70#-&#-6(&17(%--.("0+:-+'4->0'&,( cycling. Last, we outline how marine viruses are an integral part of the reef system and suggest $4&$($4-(01.,/-1'-(+.(80%/#-#(+1(%--.(./1'$0+1(0#(&1(-##-1$0&,('+>3+1-1$(+.($4-#-(:,+"&,,9( 0>3+%$&1$(-180%+1>-1$#; To p - d ow n e f f e c t s Viruses infect all cellular life, including bacteria and evidence that macroorganisms play important parts in The ecological concept that eukaryotes, and contain ~200 megatonnes of carbon the dynamics of viroplankton; for example, sponges can organismal growth and globally1 — thus, they are integral parts of marine eco- filter and consume viruses6,7. -

Discovering Viral Genomes in Human Metagenomic Data by Predicting
www.nature.com/scientificreports OPEN Discovering viral genomes in human metagenomic data by predicting unknown protein Received: 10 May 2017 Accepted: 28 November 2017 families Published online: 08 January 2018 Mauricio Barrientos-Somarribas1, David N. Messina 2, Christian Pou1, Fredrik Lysholm1,3, Annelie Bjerkner4, Tobias Allander4, Björn Andersson 1 & Erik L. L. Sonnhammer2 Massive amounts of metagenomics data are currently being produced, and in all such projects a sizeable fraction of the resulting data shows no or little homology to known sequences. It is likely that this fraction contains novel viruses, but identifcation is challenging since they frequently lack homology to known viruses. To overcome this problem, we developed a strategy to detect ORFan protein families in shotgun metagenomics data, using similarity-based clustering and a set of flters to extract bona fde protein families. We applied this method to 17 virus-enriched libraries originating from human nasopharyngeal aspirates, serum, feces, and cerebrospinal fuid samples. This resulted in 32 predicted putative novel gene families. Some families showed detectable homology to sequences in metagenomics datasets and protein databases after reannotation. Notably, one predicted family matches an ORF from the highly variable Torque Teno virus (TTV). Furthermore, follow-up from a predicted ORFan resulted in the complete reconstruction of a novel circular genome. Its organisation suggests that it most likely corresponds to a novel bacteriophage in the microviridae family, hence it was named bacteriophage HFM. Characterization of the human virome is crucial for our understanding of the role of the microbiome in health and disease. Te shif from culture-based methods to metagenomics in recent years, combined with the devel- opment of virus particle enrichment protocols, has made it possible to efciently study the entire fora of human viruses and bacteriophages associated with the human microbiome. -

Archaeal Viruses and Bacteriophages: Comparisons and Contrasts
Review Archaeal viruses and bacteriophages: comparisons and contrasts Maija K. Pietila¨ , Tatiana A. Demina, Nina S. Atanasova, Hanna M. Oksanen, and Dennis H. Bamford Institute of Biotechnology and Department of Biosciences, P.O. Box 56, Viikinkaari 5, 00014 University of Helsinki, Helsinki, Finland Isolated archaeal viruses comprise only a few percent of Euryarchaeaota [9,10]. Archaea have also been cultivated all known prokaryotic viruses. Thus, the study of viruses from moderate environments such as seawater and soil. infecting archaea is still in its early stages. Here we Consequently, an additional phylum, Thaumarchaeota, summarize the most recent discoveries of archaeal vi- has been formed to contain mesophilic and thermophilic ruses utilizing a virion-centered view. We describe the ammonia-oxidizing archaea [11]. However, all known ar- known archaeal virion morphotypes and compare them chaeal viruses infect extremophiles – mainly hyperther- to the bacterial counterparts, if such exist. Viruses infect- mophiles belonging to the crenarchaeal genera Sulfolobus ing archaea are morphologically diverse and present and Acidianus or halophiles of the euryarchaeal genera some unique morphotypes. Although limited in isolate Haloarcula, Halorubrum, and Halobacterium [6,7]. Even number, archaeal viruses reveal new insights into the though bacteria are also found in diverse extreme habitats viral world, such as deep evolutionary relationships such as hypersaline lakes, archaea typically dominate at between viruses that infect hosts from all three domains extreme salinities, based on both cultivation-dependent of life. and -independent studies [6,12–15]. Consequently, archae- al viruses do the same in hypersaline environments. About Discovery of archaeal viruses 50 prokaryotic haloviruses were recently isolated from All cellular organisms are susceptible to viral infections, nine globally distant locations, and only four of them which makes viruses a major evolutionary force shaping infected bacteria [6,16]. -

Evaluation of the Genomic Diversity of Viruses Infecting Bacteria, Archaea and Eukaryotes Using a Common Bioinformatic Platform: Steps Towards a Unified Taxonomy
RESEARCH ARTICLE Aiewsakun et al., Journal of General Virology 2018;99:1331–1343 DOI 10.1099/jgv.0.001110 Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: steps towards a unified taxonomy Pakorn Aiewsakun,1,2 Evelien M. Adriaenssens,3 Rob Lavigne,4 Andrew M. Kropinski5 and Peter Simmonds1,* Abstract Genome Relationship Applied to Virus Taxonomy (GRAViTy) is a genetics-based tool that computes sequence relatedness between viruses. Composite generalized Jaccard (CGJ) distances combine measures of homology between encoded viral genes and similarities in genome organizational features (gene orders and orientations). This scoring framework effectively recapitulates the current, largely morphology and phenotypic-based, family-level classification of eukaryotic viruses. Eukaryotic virus families typically formed monophyletic groups with consistent CGJ distance cut-off dividing between and within family divergence ranges. In the current study, a parallel analysis of prokaryotic virus families revealed quite different sequence relationships, particularly those of tailed phage families (Siphoviridae, Myoviridae and Podoviridae), where members of the same family were generally far more divergent and often not detectably homologous to each other. Analysis of the 20 currently classified prokaryotic virus families indeed split them into 70 separate clusters of tailed phages genetically equivalent to family-level assignments of eukaryotic viruses. It further divided several bacterial (Sphaerolipoviridae, Tectiviridae) and archaeal (Lipothrixviridae) families. We also found that the subfamily-level groupings of tailed phages were generally more consistent with the family assignments of eukaryotic viruses, and this supports ongoing reclassifications, including Spounavirinae and Vi1virus taxa as new virus families. The current study applied a common benchmark with which to compare taxonomies of eukaryotic and prokaryotic viruses. -
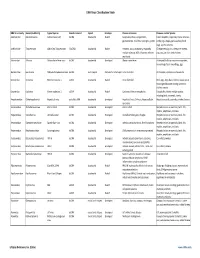
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae