The Intestinal Virome of Malabsorption Syndrome-Affected and Unaffected
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evidence for Viral Infection in the Copepods Labidocera Aestiva And
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2012 Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida Darren Stephenson Dunlap University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the American Studies Commons, Other Oceanography and Atmospheric Sciences and Meteorology Commons, and the Virology Commons Scholar Commons Citation Dunlap, Darren Stephenson, "Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida" (2012). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4032 This Thesis is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Evidence of Viruses in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida By Darren S. Dunlap A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science College of Marine Science University of South Florida Major Professor: Mya Breitbart, Ph.D Kendra Daly, Ph.D Ian Hewson, Ph.D Date of Approval: March 19, 2012 Key Words: Copepods, Single-stranded DNA Viruses, Mesozooplankton, Transmission Electron Microscopy, Metagenomics Copyright © 2012, Darren Stephenson Dunlap DEDICATION None of this would have been possible without the generous love and support of my entire family over the years. My parents, Steve and Jill Dunlap, have always encouraged my pursuits with support and love, and their persistence of throwing me into lakes and rivers is largely responsible for my passion for Marine Science. -

Nucleotide Amino Acid Size (Nt) #Orfs Marnavirus Heterosigma Akashiwo Heterosigma Akashiwo RNA Heterosigma Lang Et Al
Supplementary Table 1: Summary of information for all viruses falling within the seven Marnaviridae genera in our analyses. Accession Genome Genus Species Virus name Strain Abbreviation Source Country Reference Nucleotide Amino acid Size (nt) #ORFs Marnavirus Heterosigma akashiwo Heterosigma akashiwo RNA Heterosigma Lang et al. , 2004; HaRNAV AY337486 AAP97137 8587 One Canada RNA virus 1 virus akashiwo Tai et al. , 2003 Marine single- ASG92540 Moniruzzaman et Classification pending Q sR OV 020 KY286100 9290 Two celled USA ASG92541 al ., 2017 eukaryotes Marine single- Moniruzzaman et Classification pending Q sR OV 041 KY286101 ASG92542 9328 One celled USA al ., 2017 eukaryotes APG78557 Classification pending Wenzhou picorna-like virus 13 WZSBei69459 KX884360 9458 One Bivalve China Shi et al ., 2016 APG78557 Classification pending Changjiang picorna-like virus 2 CJLX30436 KX884547 APG79001 7171 One Crayfish China Shi et al ., 2016 Beihai picorna-like virus 57 BHHQ57630 KX883356 APG76773 8518 One Tunicate China Shi et al ., 2016 Classification pending Beihai picorna-like virus 57 BHJP51916 KX883380 APG76812 8518 One Tunicate China Shi et al ., 2016 Marine single- ASG92530 Moniruzzaman et Classification pending N OV 137 KY130494 7746 Two celled USA ASG92531 al ., 2017 eukaryotes Hubei picorna-like virus 7 WHSF7327 KX884284 APG78434 9614 One Pill worm China Shi et al ., 2016 Classification pending Hubei picorna-like virus 7 WHCC111241 KX884268 APG78407 7945 One Insect China Shi et al ., 2016 Sanxia atyid shrimp virus 2 WHCCII13331 KX884278 APG78424 10445 One Insect China Shi et al ., 2016 Classification pending Freshwater atyid Sanxia atyid shrimp virus 2 SXXX37884 KX883708 APG77465 10400 One China Shi et al ., 2016 shrimp Labyrnavirus Aurantiochytrium single Aurantiochytrium single stranded BAE47143 Aurantiochytriu AuRNAV AB193726 9035 Three4 Japan Takao et al. -

Pdf Available
Virology 554 (2021) 89–96 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/virology Diverse cressdnaviruses and an anellovirus identifiedin the fecal samples of yellow-bellied marmots Anthony Khalifeh a, Daniel T. Blumstein b,**, Rafaela S. Fontenele a, Kara Schmidlin a, C´ecile Richet a, Simona Kraberger a, Arvind Varsani a,c,* a The Biodesign Center for Fundamental and Applied Microbiomics, School of Life Sciences, Center for Evolution and Medicine, Arizona State University, Tempe, AZ, 85287, USA b Department of Ecology & Evolutionary Biology, Institute of the Environment & Sustainability, University of California Los Angeles, Los Angeles, CA, 90095, USA c Structural Biology Research Unit, Department of Clinical Laboratory Sciences, University of Cape Town, 7925, Cape Town, South Africa ARTICLE INFO ABSTRACT Keywords: Over that last decade, coupling multiple strand displacement approaches with high throughput sequencing have Marmota flaviventer resulted in the identification of genomes of diverse groups of small circular DNA viruses. Using a similar Anelloviridae approach but with recovery of complete genomes by PCR, we identified a diverse group of single-stranded vi Genomoviridae ruses in yellow-bellied marmot (Marmota flaviventer) fecal samples. From 13 fecal samples we identified viruses Cressdnaviricota in the family Genomoviridae (n = 7) and Anelloviridae (n = 1), and several others that ware part of the larger Cressdnaviricota phylum but not within established families (n = 19). There were also circular DNA molecules identified (n = 4) that appear to encode one viral-like gene and have genomes of <1545 nts. This study gives a snapshot of viruses associated with marmots based on fecal sampling. -

Multiple Origins of Prokaryotic and Eukaryotic Single-Stranded DNA Viruses from Bacterial and Archaeal Plasmids
ARTICLE https://doi.org/10.1038/s41467-019-11433-0 OPEN Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids Darius Kazlauskas 1, Arvind Varsani 2,3, Eugene V. Koonin 4 & Mart Krupovic 5 Single-stranded (ss) DNA viruses are a major component of the earth virome. In particular, the circular, Rep-encoding ssDNA (CRESS-DNA) viruses show high diversity and abundance 1234567890():,; in various habitats. By combining sequence similarity network and phylogenetic analyses of the replication proteins (Rep) belonging to the HUH endonuclease superfamily, we show that the replication machinery of the CRESS-DNA viruses evolved, on three independent occa- sions, from the Reps of bacterial rolling circle-replicating plasmids. The CRESS-DNA viruses emerged via recombination between such plasmids and cDNA copies of capsid genes of eukaryotic positive-sense RNA viruses. Similarly, the rep genes of prokaryotic DNA viruses appear to have evolved from HUH endonuclease genes of various bacterial and archaeal plasmids. Our findings also suggest that eukaryotic polyomaviruses and papillomaviruses with dsDNA genomes have evolved via parvoviruses from CRESS-DNA viruses. Collectively, our results shed light on the complex evolutionary history of a major class of viruses revealing its polyphyletic origins. 1 Institute of Biotechnology, Life Sciences Center, Vilnius University, Saulėtekio av. 7, Vilnius 10257, Lithuania. 2 The Biodesign Center for Fundamental and Applied Microbiomics, School of Life Sciences, Center for Evolution and Medicine, Arizona State University, Tempe, AZ 85287, USA. 3 Structural Biology Research Unit, Department of Integrative Biomedical Sciences, University of Cape Town, Rondebosch, 7700 Cape Town, South Africa. -

Small Circular DNA Viruses: the Muddy Viral “Playground” of Recombinant, Reassortant, and Highly Diverse Viruses
Small Circular DNA Viruses: The muddy viral “playground” of recombinant, reassortant, and highly diverse viruses @Varsani_lab Arizona State University, USA University of Canterbury, New Zealand University of Cape Town, South Africa Arvind Varsani Nucleotide substitutions Recombination Reassortment Viral evolution Viral Nucleotide substitutions Recombination Reassortment Viral evolution Viral Nucleotide substitutions Recombination Reassortment Viral evolution Viral I II III IV V dsDNA ssDNA VI VII dsRNA ssRNA(+) ssRNA(-) ssRNA(RT) dsDNA(RT) viruses Archaea Bacteria Diatoms Fungi Invertebrates Plants Vertebrates Unknown Anelloviridae – circular (~2.1 - 3.8kb) Bacillidnaviridae - circular (~4.5 - 6kb) Circoviridae – circular (~1.6 - 2.3kb) Geminiviridae – circular (~2.7kb; monopartite, bipartite, associated with α/β satellites) Genomovirdae – circular (1.8 -2.3 kb) ssDNA Inoviridae – circular (~5.8 - 8.7kb) Microviridae – circular (~4.5 - 6kb) Nanoviridae – circular (~1.2kb; multicomponent viruses; associated with satellites) Parvoviridae – linear (~3.7 - 6kb) Pleolipoviridae* – circular ssDNA; circular dsDNA or liner dsDNA (7-16kb) Smacoviridae* - circular (2.3 - 2.9kb) ssDNA viruses Chimerism in Rep Geminiviruses MSV: ~2 x 10-4 subs/site/year SSRV: ~3 x 10-4 subs/site/year EACMV: ~1 x 10-3 subs/site/year TYLCV: ~3 x 10-4 subs/site/year Nanoviruses FBNYV: ~1 x 10-3 subs/site/year BBTV: ~3 x 10-4 subs/site/year Circoviruses BFDV: ~2 x 10-3 subs/site/year PCV-2: ~1 x 10-3 subs/site/year Parvoviruses CPV: ~2 x 10-4 - 8 x 10-5 subs/site/year -

First Description of Adenovirus, Enterovirus, Rotavirus and Torque
First description of Adenovirus, Enterovirus, Rotavirus and Torque teno virus in water samples collected from the Arroio Dilúvio, Porto Alegre, Brazil Vecchia, AD.a,b, Fleck, JD.a,b, Comerlato, J.c, Kluge, M.b, Bergamaschi, B.c, Da Silva, JVS.b, Da Luz, RB.b, Teixeira, TF.b, Garbinatto, GN.d, Oliveira, DV.d, Zanin, JG.d, Van der Sand, S.d, Frazzon, APG.d, Franco, AC.c, Roehe, PM.c,e and Spilki, FR.a,b* aPrograma de Pós-Graduação em Qualidade Ambiental, Universidade Feevale, CEP 93352-000, Novo Hamburgo, RS, Brazil bLaboratório de Microbiologia Molecular, Instituto de Ciências da Saúde, Universidade Feevale, CEP 93352-000, Novo Hamburgo, RS, Brazil cLaboratório de Virologia, Departamento de Microbiologia, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul – UFRGS, Av. Sarmento Leite, 500, CEP 90050-170, Porto Alegre, RS, Brazil dDepartamento de Microbiologia, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul – UFRGS, Av. Sarmento Leite, 500, CEP 90050-170, Porto Alegre, RS, Brazil eInstituto de Pesquisa Veterinária “Desidério Finamor” – IPVDF, Fundação Estadual de Pesquisa Agropecuária – FEPAGRO-Saúde Animal, Estrada do Conde, 6000, CEP 92990-000, Eldorado do Sul, RS, Brazil *e-mail: [email protected] Received May 11, 2011 – Accepted July 14, 2011 – Distributed May 31, 2012 (With 1 figure) Abstract Adenovirus (AdV), enterovirus (EV), genogroup A rotaviruses (GARV) and Torque teno virus (TTV) are non-enveloped viral agents excreted in feces and so may contaminate water bodies. In the present study, the molecular detection of these viruses was performed in samples of surface water collected from the Arroio Dilúvio, a waterstream that crosses the city of Porto Alegre, RS, Brazil, receiving great volumes of non-treated sewage from a large urban area. -

The Viruses of Wild Pigeon Droppings
The Viruses of Wild Pigeon Droppings Tung Gia Phan1,2, Nguyen Phung Vo1,3,A´ kos Boros4,Pe´ter Pankovics4,Ga´bor Reuter4, Olive T. W. Li6, Chunling Wang5, Xutao Deng1, Leo L. M. Poon6, Eric Delwart1,2* 1 Blood Systems Research Institute, San Francisco, California, United States of America, 2 Department of Laboratory Medicine, University of California San Francisco, San Francisco, California, United States of America, 3 Pharmacology Department, School of Pharmacy, Ho Chi Minh City University of Medicine and Pharmacy, Ho Chi Minh, Vietnam, 4 Regional Laboratory of Virology, National Reference Laboratory of Gastroenteric Viruses, A´ NTSZ Regional Institute of State Public Health Service, Pe´cs, Hungary, 5 Stanford Genome Technology Center, Stanford, California, United States of America, 6 Centre of Influenza Research and School of Public Health, University of Hong Kong, Hong Kong SAR Abstract Birds are frequent sources of emerging human infectious diseases. Viral particles were enriched from the feces of 51 wild urban pigeons (Columba livia) from Hong Kong and Hungary, their nucleic acids randomly amplified and then sequenced. We identified sequences from known and novel species from the viral families Circoviridae, Parvoviridae, Picornaviridae, Reoviridae, Adenovirus, Astroviridae, and Caliciviridae (listed in decreasing number of reads), as well as plant and insect viruses likely originating from consumed food. The near full genome of a new species of a proposed parvovirus genus provisionally called Aviparvovirus contained an unusually long middle ORF showing weak similarity to an ORF of unknown function from a fowl adenovirus. Picornaviruses found in both Asia and Europe that are distantly related to the turkey megrivirus and contained a highly divergent 2A1 region were named mesiviruses. -

Computational Exploration of Virus Diversity on Transcriptomic Datasets
Computational Exploration of Virus Diversity on Transcriptomic Datasets Digitaler Anhang der Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn vorgelegt von Simon Käfer aus Andernach Bonn 2019 Table of Contents 1 Table of Contents 1 Preliminary Work - Phylogenetic Tree Reconstruction 3 1.1 Non-segmented RNA Viruses ........................... 3 1.2 Segmented RNA Viruses ............................. 4 1.3 Flavivirus-like Superfamily ............................ 5 1.4 Picornavirus-like Viruses ............................. 6 1.5 Togavirus-like Superfamily ............................ 7 1.6 Nidovirales-like Viruses .............................. 8 2 TRAVIS - True Positive Details 9 2.1 INSnfrTABRAAPEI-14 .............................. 9 2.2 INSnfrTADRAAPEI-16 .............................. 10 2.3 INSnfrTAIRAAPEI-21 ............................... 11 2.4 INSnfrTAORAAPEI-35 .............................. 13 2.5 INSnfrTATRAAPEI-43 .............................. 14 2.6 INSnfrTBERAAPEI-19 .............................. 15 2.7 INSytvTABRAAPEI-11 .............................. 16 2.8 INSytvTALRAAPEI-35 .............................. 17 2.9 INSytvTBORAAPEI-47 .............................. 18 2.10 INSswpTBBRAAPEI-21 .............................. 19 2.11 INSeqtTAHRAAPEI-88 .............................. 20 2.12 INShkeTCLRAAPEI-44 .............................. 22 2.13 INSeqtTBNRAAPEI-11 .............................. 23 2.14 INSeqtTCJRAAPEI-20 -

Viral Diversity in Tree Species
Universidade de Brasília Instituto de Ciências Biológicas Departamento de Fitopatologia Programa de Pós-Graduação em Biologia Microbiana Doctoral Thesis Viral diversity in tree species FLÁVIA MILENE BARROS NERY Brasília - DF, 2020 FLÁVIA MILENE BARROS NERY Viral diversity in tree species Thesis presented to the University of Brasília as a partial requirement for obtaining the title of Doctor in Microbiology by the Post - Graduate Program in Microbiology. Advisor Dra. Rita de Cássia Pereira Carvalho Co-advisor Dr. Fernando Lucas Melo BRASÍLIA, DF - BRAZIL FICHA CATALOGRÁFICA NERY, F.M.B Viral diversity in tree species Flávia Milene Barros Nery Brasília, 2025 Pages number: 126 Doctoral Thesis - Programa de Pós-Graduação em Biologia Microbiana, Universidade de Brasília, DF. I - Virus, tree species, metagenomics, High-throughput sequencing II - Universidade de Brasília, PPBM/ IB III - Viral diversity in tree species A minha mãe Ruth Ao meu noivo Neil Dedico Agradecimentos A Deus, gratidão por tudo e por ter me dado uma família e amigos que me amam e me apoiam em todas as minhas escolhas. Minha mãe Ruth e meu noivo Neil por todo o apoio e cuidado durante os momentos mais difíceis que enfrentei durante minha jornada. Aos meus irmãos André, Diego e meu sobrinho Bruno Kawai, gratidão. Aos meus amigos de longa data Rafaelle, Evanessa, Chênia, Tati, Leo, Suzi, Camilets, Ricardito, Jorgito e Diego, saudade da nossa amizade e dos bons tempos. Amo vocês com todo o meu coração! Minha orientadora e grande amiga Profa Rita de Cássia Pereira Carvalho, a quem escolhi e fui escolhida para amar e fazer parte da família. -
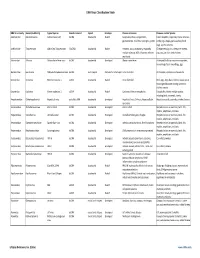
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae -

Viruses in Transplantation - Not Always Enemies
Viruses in transplantation - not always enemies Virome and transplantation ECCMID 2018 - Madrid Prof. Laurent Kaiser Head Division of Infectious Diseases Laboratory of Virology Geneva Center for Emerging Viral Diseases University Hospital of Geneva ESCMID eLibrary © by author Conflict of interest None ESCMID eLibrary © by author The human virome: definition? Repertoire of viruses found on the surface of/inside any body fluid/tissue • Eukaryotic DNA and RNA viruses • Prokaryotic DNA and RNA viruses (phages) 25 • The “main” viral community (up to 10 bacteriophages in humans) Haynes M. 2011, Metagenomic of the human body • Endogenous viral elements integrated into host chromosomes (8% of the human genome) • NGS is shaping the definition Rascovan N et al. Annu Rev Microbiol 2016;70:125-41 Popgeorgiev N et al. Intervirology 2013;56:395-412 Norman JM et al. Cell 2015;160:447-60 ESCMID eLibraryFoxman EF et al. Nat Rev Microbiol 2011;9:254-64 © by author Viruses routinely known to cause diseases (non exhaustive) Upper resp./oropharyngeal HSV 1 Influenza CNS Mumps virus Rhinovirus JC virus RSV Eye Herpes viruses Parainfluenza HSV Measles Coronavirus Adenovirus LCM virus Cytomegalovirus Flaviviruses Rabies HHV6 Poliovirus Heart Lower respiratory HTLV-1 Coxsackie B virus Rhinoviruses Parainfluenza virus HIV Coronaviruses Respiratory syncytial virus Parainfluenza virus Adenovirus Respiratory syncytial virus Coronaviruses Gastro-intestinal Influenza virus type A and B Human Bocavirus 1 Adenovirus Hepatitis virus type A, B, C, D, E Those that cause -

Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics
viruses Article Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics Ashleigh F. Porter 1, Mang Shi 1, John-Sebastian Eden 1,2 , Yong-Zhen Zhang 3,4 and Edward C. Holmes 1,3,* 1 Marie Bashir Institute for Infectious Diseases and Biosecurity, Charles Perkins Centre, School of Life & Environmental Sciences and Sydney Medical School, The University of Sydney, Sydney, NSW 2006, Australia; [email protected] (A.F.P.); [email protected] (M.S.); [email protected] (J.-S.E.) 2 Centre for Virus Research, Westmead Institute for Medical Research, Westmead, NSW 2145, Australia 3 Shanghai Public Health Clinical Center and School of Public Health, Fudan University, Shanghai 201500, China; [email protected] 4 Department of Zoonosis, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Changping, Beijing 102206, China * Correspondence: [email protected]; Tel.: +61-2-9351-5591 Received: 17 October 2019; Accepted: 23 November 2019; Published: 25 November 2019 Abstract: DNA viruses comprise a wide array of genome structures and infect diverse host species. To date, most studies of DNA viruses have focused on those with the strongest disease associations. Accordingly, there has been a marked lack of sampling of DNA viruses from invertebrates. Bulk RNA sequencing has resulted in the discovery of a myriad of novel RNA viruses, and herein we used this methodology to identify actively transcribing DNA viruses in meta-transcriptomic libraries of diverse invertebrate species. Our analysis revealed high levels of phylogenetic diversity in DNA viruses, including 13 species from the Parvoviridae, Circoviridae, and Genomoviridae families of single-stranded DNA virus families, and six double-stranded DNA virus species from the Nudiviridae, Polyomaviridae, and Herpesviridae, for which few invertebrate viruses have been identified to date.