Intestinal Virome Changes Precede Autoimmunity in Type I Diabetes-Susceptible Children,” by Guoyan Zhao, Tommi Vatanen, Lindsay Droit, Arnold Park, Aleksandar D
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Elisabeth Mendes Martins De Moura Diversidade De Vírus DNA
Elisabeth Mendes Martins de Moura Diversidade de vírus DNA autóctones e alóctones de mananciais e de esgoto da região metropolitana de São Paulo Tese apresentada ao Programa de Pós- Graduação em Microbiologia do Instituto de Ciências Biomédicas da Universidade de São Paulo, para obtenção do Titulo de Doutor em Ciências. Área de concentração: Microbiologia Orienta: Prof (a). Dr (a). Dolores Ursula Mehnert versão original São Paulo 2017 RESUMO MOURA, E. M. M. Diversidade de vírus DNA autóctones e alóctones de mananciais e de esgoto da região metropolitana de São Paulo. 2017. 134f. Tese (Doutorado em Microbiologia) - Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, 2017. A água doce no Brasil, assim como o seu consumo é extremamente importante para as diversas atividades criadas pelo ser humano. Por esta razão o consumo deste bem é muito grande e consequentemente, provocando o seu impacto. Os mananciais são normalmente usados para abastecimento doméstico, comercial, industrial e outros fins. Os estudos na área de ecologia de micro-organismos nos ecossistemas aquáticos (mananciais) e em esgotos vêm sendo realizados com mais intensidade nos últimos anos. Nas últimas décadas foi introduzido o conceito de virioplâncton com base na abundância e diversidade de partículas virais presentes no ambiente aquático. O virioplâncton influencia muitos processos ecológicos e biogeoquímicos, como ciclagem de nutriente, taxa de sedimentação de partículas, diversidade e distribuição de espécies de algas e bactérias, controle de florações de fitoplâncton e transferência genética horizontal. Os estudos nesta área da virologia molecular ainda estão muito restritos no país, bem como muito pouco se conhece sobre a diversidade viral na água no Brasil. -

Changes to Virus Taxonomy 2004
Arch Virol (2005) 150: 189–198 DOI 10.1007/s00705-004-0429-1 Changes to virus taxonomy 2004 M. A. Mayo (ICTV Secretary) Scottish Crop Research Institute, Invergowrie, Dundee, U.K. Received July 30, 2004; accepted September 25, 2004 Published online November 10, 2004 c Springer-Verlag 2004 This note presents a compilation of recent changes to virus taxonomy decided by voting by the ICTV membership following recommendations from the ICTV Executive Committee. The changes are presented in the Table as decisions promoted by the Subcommittees of the EC and are grouped according to the major hosts of the viruses involved. These new taxa will be presented in more detail in the 8th ICTV Report scheduled to be published near the end of 2004 (Fauquet et al., 2004). Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., and Ball, L.A. (eds) (2004). Virus Taxonomy, VIIIth Report of the ICTV. Elsevier/Academic Press, London, pp. 1258. Recent changes to virus taxonomy Viruses of vertebrates Family Arenaviridae • Designate Cupixi virus as a species in the genus Arenavirus • Designate Bear Canyon virus as a species in the genus Arenavirus • Designate Allpahuayo virus as a species in the genus Arenavirus Family Birnaviridae • Assign Blotched snakehead virus as an unassigned species in family Birnaviridae Family Circoviridae • Create a new genus (Anellovirus) with Torque teno virus as type species Family Coronaviridae • Recognize a new species Severe acute respiratory syndrome coronavirus in the genus Coro- navirus, family Coronaviridae, order Nidovirales -

Evidence for Viral Infection in the Copepods Labidocera Aestiva And
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2012 Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida Darren Stephenson Dunlap University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the American Studies Commons, Other Oceanography and Atmospheric Sciences and Meteorology Commons, and the Virology Commons Scholar Commons Citation Dunlap, Darren Stephenson, "Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida" (2012). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4032 This Thesis is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Evidence of Viruses in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida By Darren S. Dunlap A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science College of Marine Science University of South Florida Major Professor: Mya Breitbart, Ph.D Kendra Daly, Ph.D Ian Hewson, Ph.D Date of Approval: March 19, 2012 Key Words: Copepods, Single-stranded DNA Viruses, Mesozooplankton, Transmission Electron Microscopy, Metagenomics Copyright © 2012, Darren Stephenson Dunlap DEDICATION None of this would have been possible without the generous love and support of my entire family over the years. My parents, Steve and Jill Dunlap, have always encouraged my pursuits with support and love, and their persistence of throwing me into lakes and rivers is largely responsible for my passion for Marine Science. -

Guide for Common Viral Diseases of Animals in Louisiana
Sampling and Testing Guide for Common Viral Diseases of Animals in Louisiana Please click on the species of interest: Cattle Deer and Small Ruminants The Louisiana Animal Swine Disease Diagnostic Horses Laboratory Dogs A service unit of the LSU School of Veterinary Medicine Adapted from Murphy, F.A., et al, Veterinary Virology, 3rd ed. Cats Academic Press, 1999. Compiled by Rob Poston Multi-species: Rabiesvirus DCN LADDL Guide for Common Viral Diseases v. B2 1 Cattle Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 2 Deer and Small Ruminants Please click on the principle system involvement Generalized viral disease Respiratory viral disease Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 3 Swine Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 4 Horses Please click on the principle system involvement Generalized viral diseases Neurological viral diseases Respiratory viral diseases Enteric viral diseases Abortifacient/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 5 Dogs Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. -

Seroprevalence of Antibodies to Primate Erythroparvovirus 1 (B19V) in Australia Helen M
Faddy et al. BMC Infectious Diseases (2018) 18:631 https://doi.org/10.1186/s12879-018-3525-7 RESEARCHARTICLE Open Access Seroprevalence of antibodies to primate erythroparvovirus 1 (B19V) in Australia Helen M. Faddy1,2* , Elise C. Gorman1,2, Veronica C. Hoad3, Francesca D. Frentiu2, Sarah Tozer4 and R. L. P. Flower1,2 Abstract Backgroud: Primate erythroparvovirus 1 (B19V) is a globally ubiquitous DNA virus. Infection results in a variety of clinical presentations including erythema infectiosum in children and arthralgia in adults. There is limited understanding of the seroprevalence of B19V antibodies in the Australian population and therefore of population- wide immunity. This study aimed to investigate the seroprevalence of B19V antibodies in an Australian blood donor cohort, along with a cohort from a paediatric population. Methods: Age/sex/geographical location stratified plasma samples (n = 2221) were collected from Australian blood donors. Samples were also sourced from paediatric patients (n = 223) in Queensland. All samples were screened for B19V IgG using an indirect- enzyme-linked immunosorbent assay. Results: Overall, 57.90% (95% CI: 55.94%–59.85%) of samples tested positive for B19V IgG, with the national age- standardized seroprevalence of B19V exposure in Australians aged 0 to 79 years estimated to be 54.41%. Increasing age (p < 0.001) and state of residence (p < 0.001) were independently associated with B19V exposure in blood donors, with the highest rates in donors from Tasmania (71.88%, 95% CI: 66.95%–76.80%) and donors aged 65–80 years (78.41%, 95% CI: 74.11%–82.71%). A seroprevalence of 52.04% (95% CI: 47.92%–56.15%) was reported in women of child-bearing age (16 to 44 years). -

Genetic Content and Evolution of Adenoviruses Andrew J
Journal of General Virology (2003), 84, 2895–2908 DOI 10.1099/vir.0.19497-0 Review Genetic content and evolution of adenoviruses Andrew J. Davison,1 Ma´ria Benko´´ 2 and Bala´zs Harrach2 Correspondence 1MRC Virology Unit, Institute of Virology, Church Street, Glasgow G11 5JR, UK Andrew Davison 2Veterinary Medical Research Institute, Hungarian Academy of Sciences, H-1581 Budapest, [email protected] Hungary This review provides an update of the genetic content, phylogeny and evolution of the family Adenoviridae. An appraisal of the condition of adenovirus genomics highlights the need to ensure that public sequence information is interpreted accurately. To this end, all complete genome sequences available have been reannotated. Adenoviruses fall into four recognized genera, plus possibly a fifth, which have apparently evolved with their vertebrate hosts, but have also engaged in a number of interspecies transmission events. Genes inherited by all modern adenoviruses from their common ancestor are located centrally in the genome and are involved in replication and packaging of viral DNA and formation and structure of the virion. Additional niche-specific genes have accumulated in each lineage, mostly near the genome termini. Capture and duplication of genes in the setting of a ‘leader–exon structure’, which results from widespread use of splicing, appear to have been central to adenovirus evolution. The antiquity of the pre-vertebrate lineages that ultimately gave rise to the Adenoviridae is illustrated by morphological similarities between adenoviruses and bacteriophages, and by use of a protein-primed DNA replication strategy by adenoviruses, certain bacteria and bacteriophages, and linear plasmids of fungi and plants. -

Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats
viruses Article Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats Laura M. Bergner 1,2,* , Nardus Mollentze 1,2 , Richard J. Orton 2 , Carlos Tello 3,4, Alice Broos 2, Roman Biek 1 and Daniel G. Streicker 1,2 1 Institute of Biodiversity, Animal Health and Comparative Medicine, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow G12 8QQ, UK; [email protected] (N.M.); [email protected] (R.B.); [email protected] (D.G.S.) 2 MRC–University of Glasgow Centre for Virus Research, Glasgow G61 1QH, UK; [email protected] (R.J.O.); [email protected] (A.B.) 3 Association for the Conservation and Development of Natural Resources, Lima 15037, Peru; [email protected] 4 Yunkawasi, Lima 15049, Peru * Correspondence: [email protected] Abstract: The contemporary surge in metagenomic sequencing has transformed knowledge of viral diversity in wildlife. However, evaluating which newly discovered viruses pose sufficient risk of infecting humans to merit detailed laboratory characterization and surveillance remains largely speculative. Machine learning algorithms have been developed to address this imbalance by ranking the relative likelihood of human infection based on viral genome sequences, but are not yet routinely Citation: Bergner, L.M.; Mollentze, applied to viruses at the time of their discovery. Here, we characterized viral genomes detected N.; Orton, R.J.; Tello, C.; Broos, A.; through metagenomic sequencing of feces and saliva from common vampire bats (Desmodus rotundus) Biek, R.; Streicker, D.G. and used these data as a case study in evaluating zoonotic potential using molecular sequencing Characterizing and Evaluating the data. -

Porcine Kobuvirus
PORCINE KOBUVIRUS Prepared for the Swine Health Information Center By the Center for Food Security and Public Health, College of Veterinary Medicine, Iowa State University September 2015 SUMMARY Etiology • Porcine kobuvirus (PKoV) is a small, non-enveloped RNA virus in the family Picornaviridae. • There are three distinct clusters within the genus Kobuvirus: Aichivirus A (AiV-A) includes human AiV-1, canine KoV-1, and murine KoV-1. Aichivirus B (AiV-B) includes bovine KoV-1 and sheep KoV-1. Aichivirus C (AiV-C) includes porcine KoV-1 (PKoV/AiV-C).1 Cleaning and Disinfection • AiV-1 is readily inactivated at 56ºC after 20 minutes. • There is no published information about the susceptibility of PKoV/AiV-C to disinfectants. Kobuviruses (KoVs) are potentially susceptible to disinfection with acids like acetic acid, aldehydes like glutaraldehyde, alkalis like sodium hydroxide, and oxidizing agents like Virkon- S®11. Epidemiology • Kobuviruses infect many different species. The AiV-C cluster contains swine viruses exclusively. • PKoV/AiV-C has been isolated from swine herds in China, Thailand, Japan, South Korea, Italy, Hungary, Czech Republic, the United States, the Netherlands, Kenya, Uganda, and Brazil. • Prevalence in domestic pigs ranges from 13–99%. One study of pigs in the United States showed that 21.7% of healthy and 21.9% of diarrheic samples were PKoV/AiV-C-positive. Transmission • Transmission is thought to be fecal-oral. • Wild boars might be a source of infection for domestic swine. Infection in Swine/Pathogenesis • PKoV/AiV-C has been implicated as the cause of an outbreak of diarrhea, dehydration, and vomiting in Chinese piglets. -

Virus–Host Interactions and Their Roles in Coral Reef Health and Disease
!"#$"%& Virus–host interactions and their roles in coral reef health and disease Rebecca Vega Thurber1, Jérôme P. Payet1,2, Andrew R. Thurber1,2 and Adrienne M. S. Correa3 !"#$%&'$()(*+%&,(%--.#(+''/%!01(1/$%0-1$23++%(#4&,,+5(5&$-%#6('+1#$0$/$-("0+708-%#0$9(&17( 3%+7/'$080$9(4+$#3+$#6(&17(&%-($4%-&$-1-7("9(&1$4%+3+:-10'(70#$/%"&1'-;(<40#(=-80-5(3%+807-#( &1(01$%+7/'$0+1($+('+%&,(%--.(80%+,+:9(&17(->34�?-#($4-(,01@#("-$5--1(80%/#-#6('+%&,(>+%$&,0$9( &17(%--.(-'+#9#$->(7-',01-;(A-(7-#'%0"-($4-(70#$01'$08-("-1$40'2&##+'0&$-7(&17(5&$-%2'+,/>12( &##+'0&$-7(80%+>-#($4&$(&%-(/10B/-($+('+%&,(%--.#6(540'4(4&8-(%-'-08-7(,-##(&$$-1$0+1($4&1( 80%/#-#(01(+3-12+'-&1(#9#$->#;(A-(493+$4-#0?-($4&$(80%/#-#(+.("&'$-%0&(&17(-/@&%9+$-#( 791&>0'&,,9(01$-%&'$(50$4($4-0%(4+#$#(01($4-(5&$-%('+,/>1(&17(50$4(#',-%&'$010&1(C#$+19D('+%&,#($+( 01.,/-1'-(>0'%+"0&,('+>>/10$9(791&>0'#6('+%&,(",-&'401:(&17(70#-&#-6(&17(%--.("0+:-+'4->0'&,( cycling. Last, we outline how marine viruses are an integral part of the reef system and suggest $4&$($4-(01.,/-1'-(+.(80%/#-#(+1(%--.(./1'$0+1(0#(&1(-##-1$0&,('+>3+1-1$(+.($4-#-(:,+"&,,9( 0>3+%$&1$(-180%+1>-1$#; To p - d ow n e f f e c t s Viruses infect all cellular life, including bacteria and evidence that macroorganisms play important parts in The ecological concept that eukaryotes, and contain ~200 megatonnes of carbon the dynamics of viroplankton; for example, sponges can organismal growth and globally1 — thus, they are integral parts of marine eco- filter and consume viruses6,7. -

Allosteric Integrase Inhibitor Potency Is Determined Through the Inhibition of HIV-1 Particle Maturation
Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation Kellie A. Juradoa, Hao Wanga, Alison Slaughterb, Lei Fengb, Jacques J. Kesslb, Yasuhiro Koha, Weifeng Wanga, Allison Ballandras-Colasa, Pratiq A. Patelc, James R. Fuchsc, Mamuka Kvaratskheliab, and Alan Engelmana,1 aDepartment of Cancer Immunology and AIDS, Dana-Farber Cancer Institute and Department of Medicine, Harvard Medical School, Boston, MA 02215; and bCenter for Retrovirus Research and Comprehensive Cancer Center and cDivision of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University, Columbus, OH 43210 Edited by Alan R. Rein, National Cancer Institute, Frederick, MD, and accepted by the Editorial Board April 1, 2013 (received for review January 14, 2013) Integration is essential for HIV-1 replication, and the viral integrase HIV-1 preferentially integrates along the bodies of active genes (IN) protein is an important therapeutic target. Allosteric IN inhib- (6), a trait that is largely attributable to an interaction between itors (ALLINIs) that engage the IN dimer interface at the binding site IN and the host protein lens epithelium-derived growth factor for the host protein lens epithelium-derived growth factor (LEDGF)/ (LEDGF)/transcriptional coactivator p75 (reviewed in refs. 7 and transcriptional coactivator p75 are an emerging class of small mole- 8). LEDGF/p75 functions as a bimodal tether during integration: cule antagonists. Consistent with the inhibition of a multivalent drug elements within its N-terminal region confer constitutive binding to target, ALLINIs display steep antiviral dose–response curves ex vivo. chromatin, whereas a downstream IN-binding domain (IBD) binds ALLINIs multimerize IN protein and concordantly block its assembly lentiviral IN proteins (9, 10). -
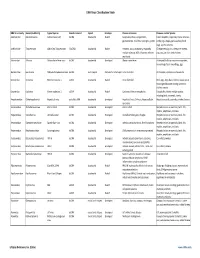
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae -

Viruses in Transplantation - Not Always Enemies
Viruses in transplantation - not always enemies Virome and transplantation ECCMID 2018 - Madrid Prof. Laurent Kaiser Head Division of Infectious Diseases Laboratory of Virology Geneva Center for Emerging Viral Diseases University Hospital of Geneva ESCMID eLibrary © by author Conflict of interest None ESCMID eLibrary © by author The human virome: definition? Repertoire of viruses found on the surface of/inside any body fluid/tissue • Eukaryotic DNA and RNA viruses • Prokaryotic DNA and RNA viruses (phages) 25 • The “main” viral community (up to 10 bacteriophages in humans) Haynes M. 2011, Metagenomic of the human body • Endogenous viral elements integrated into host chromosomes (8% of the human genome) • NGS is shaping the definition Rascovan N et al. Annu Rev Microbiol 2016;70:125-41 Popgeorgiev N et al. Intervirology 2013;56:395-412 Norman JM et al. Cell 2015;160:447-60 ESCMID eLibraryFoxman EF et al. Nat Rev Microbiol 2011;9:254-64 © by author Viruses routinely known to cause diseases (non exhaustive) Upper resp./oropharyngeal HSV 1 Influenza CNS Mumps virus Rhinovirus JC virus RSV Eye Herpes viruses Parainfluenza HSV Measles Coronavirus Adenovirus LCM virus Cytomegalovirus Flaviviruses Rabies HHV6 Poliovirus Heart Lower respiratory HTLV-1 Coxsackie B virus Rhinoviruses Parainfluenza virus HIV Coronaviruses Respiratory syncytial virus Parainfluenza virus Adenovirus Respiratory syncytial virus Coronaviruses Gastro-intestinal Influenza virus type A and B Human Bocavirus 1 Adenovirus Hepatitis virus type A, B, C, D, E Those that cause