The Discovery, Distribution and Diversity of DNA Viruses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Using Cell Lines to Study Factors Affecting Transmission of Fish Viruses
Using cell lines to study factors affecting transmission of fish viruses by Phuc Hoang Pham A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Doctor of Philosophy in Biology Waterloo, Ontario, Canada, 2014 ©Phuc Hoang Pham 2014 AUTHOR'S DECLARATION I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii ABSTRACT Factors that can influence the transmission of aquatic viruses in fish production facilities and natural environment are the immune defense of host species, the ability of viruses to infect host cells, and the environmental persistence of viruses. In this thesis, fish cell lines were used to study different aspects of these factors. Five viruses were used in this study: viral hemorrhagic septicemia virus (VHSV) from the Rhabdoviridae family; chum salmon reovirus (CSV) from the Reoviridae family; infectious pancreatic necrosis virus (IPNV) from the Birnaviridae family; and grouper iridovirus (GIV) and frog virus-3 (FV3) from the Iridoviridae family. The first factor affecting the transmission of fish viruses examined in this thesis is the immune defense of host species. In this work, infections of marine VHSV-IVa and freshwater VHSV-IVb were studied in two rainbow trout cell lines, RTgill-W1 from the gill epithelium, and RTS11 from spleen macrophages. RTgill-W1 produced infectious progeny of both VHSV-IVa and -IVb. However, VHSV-IVa was more infectious than IVb toward RTgill-W1: IVa caused cytopathic effects (CPE) at a lower viral titre, elicited CPE earlier, and yielded higher titres. -

Changes to Virus Taxonomy 2004
Arch Virol (2005) 150: 189–198 DOI 10.1007/s00705-004-0429-1 Changes to virus taxonomy 2004 M. A. Mayo (ICTV Secretary) Scottish Crop Research Institute, Invergowrie, Dundee, U.K. Received July 30, 2004; accepted September 25, 2004 Published online November 10, 2004 c Springer-Verlag 2004 This note presents a compilation of recent changes to virus taxonomy decided by voting by the ICTV membership following recommendations from the ICTV Executive Committee. The changes are presented in the Table as decisions promoted by the Subcommittees of the EC and are grouped according to the major hosts of the viruses involved. These new taxa will be presented in more detail in the 8th ICTV Report scheduled to be published near the end of 2004 (Fauquet et al., 2004). Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., and Ball, L.A. (eds) (2004). Virus Taxonomy, VIIIth Report of the ICTV. Elsevier/Academic Press, London, pp. 1258. Recent changes to virus taxonomy Viruses of vertebrates Family Arenaviridae • Designate Cupixi virus as a species in the genus Arenavirus • Designate Bear Canyon virus as a species in the genus Arenavirus • Designate Allpahuayo virus as a species in the genus Arenavirus Family Birnaviridae • Assign Blotched snakehead virus as an unassigned species in family Birnaviridae Family Circoviridae • Create a new genus (Anellovirus) with Torque teno virus as type species Family Coronaviridae • Recognize a new species Severe acute respiratory syndrome coronavirus in the genus Coro- navirus, family Coronaviridae, order Nidovirales -

Evidence for Viral Infection in the Copepods Labidocera Aestiva And
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2012 Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida Darren Stephenson Dunlap University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the American Studies Commons, Other Oceanography and Atmospheric Sciences and Meteorology Commons, and the Virology Commons Scholar Commons Citation Dunlap, Darren Stephenson, "Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida" (2012). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4032 This Thesis is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Evidence of Viruses in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida By Darren S. Dunlap A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science College of Marine Science University of South Florida Major Professor: Mya Breitbart, Ph.D Kendra Daly, Ph.D Ian Hewson, Ph.D Date of Approval: March 19, 2012 Key Words: Copepods, Single-stranded DNA Viruses, Mesozooplankton, Transmission Electron Microscopy, Metagenomics Copyright © 2012, Darren Stephenson Dunlap DEDICATION None of this would have been possible without the generous love and support of my entire family over the years. My parents, Steve and Jill Dunlap, have always encouraged my pursuits with support and love, and their persistence of throwing me into lakes and rivers is largely responsible for my passion for Marine Science. -

Characterization of Viral Communities of Biting Midges and Identification
viruses Article Characterization of Viral Communities of Biting Midges and Identification of Novel Thogotovirus Species and Rhabdovirus Genus Sarah Temmam 1, Sonia Monteil-Bouchard 1, Catherine Robert 1, Jean-Pierre Baudoin 1, Masse Sambou 1, Maxence Aubadie-Ladrix 1, Noémie Labas 1, Didier Raoult 1,2, Oleg Mediannikov 1 and Christelle Desnues 1,* 1 Unité de Recherche sur les Maladies Infectieuses et Tropicales Emergentes (URMITE), UM63 CNRS 7278 IRD 198 INSERM U1095, Aix-Marseille Université, Marseille 13005, France; [email protected] (S.T.); [email protected] (S.M.-B.); [email protected] (C.R.); [email protected] (J.-P.B.); [email protected] (M.S.); [email protected] (M.A.-L.); [email protected] (N.L.); [email protected] (D.R.); [email protected] (O.M.). 2 Fondation IHU Méditerranée Infection, Pôle des Maladies Infectieuses et Tropicales Clinique et Biologique, Fédération de Bactériologie-Hygiène-Virologie, Centre Hospitalo-Universitaire Timone, Méditerranée Infection, Assistance Publique–Hôpitaux de Marseille, Marseille 13005, France * Correspondence: [email protected]; Tel.: +33-0-491324630; Fax: +33-0-491387772 Academic Editors: Johnson Mak, Peter Walker and Marcus Thomas Gilbert Received: 21 October 2015; Accepted: 1 March 2016; Published: 11 March 2016 Abstract: More than two thirds of emerging viruses are of zoonotic origin, and among them RNA viruses represent the majority. Ceratopogonidae (genus Culicoides) are well-known vectors of several viruses responsible for epizooties (bluetongue, epizootic haemorrhagic disease, etc.). They are also vectors of the only known virus infecting humans: the Oropouche virus. Female midges usually feed on a variety of hosts, leading to possible transmission of emerging viruses from animals to humans. -

Characterization of Farmington Virus, a Novel Virus from Birds That Is Distantly Related to Members of the Family Rhabdoviridae
Palacios et al. Virology Journal 2013, 10:219 http://www.virologyj.com/content/10/1/219 RESEARCH Open Access Characterization of Farmington virus, a novel virus from birds that is distantly related to members of the family Rhabdoviridae Gustavo Palacios1†, Naomi L Forrester2,3,4†, Nazir Savji5,7†, Amelia P A Travassos da Rosa2, Hilda Guzman2, Kelly DeToy5, Vsevolod L Popov2,4, Peter J Walker6, W Ian Lipkin5, Nikos Vasilakis2,3,4 and Robert B Tesh2,4* Abstract Background: Farmington virus (FARV) is a rhabdovirus that was isolated from a wild bird during an outbreak of epizootic eastern equine encephalitis on a pheasant farm in Connecticut, USA. Findings: Analysis of the nearly complete genome sequence of the prototype CT AN 114 strain indicates that it encodes the five canonical rhabdovirus structural proteins (N, P, M, G and L) with alternative ORFs (> 180 nt) in the N and G genes. Phenotypic and genetic characterization of FARV has confirmed that it is a novel rhabdovirus and probably represents a new species within the family Rhabdoviridae. Conclusions: In sum, our analysis indicates that FARV represents a new species within the family Rhabdoviridae. Keywords: Farmington virus (FARV), Family Rhabdoviridae, Next generation sequencing, Phylogeny Background Results Therhabdovirusesarealargeanddiversegroupofsingle- Growth characteristics stranded, negative sense RNA viruses that infect a wide Three litters of newborn (1–2 day old) ICR mice with aver- range of vertebrates, invertebrates and plants [1]. The family agesizeof10pupswereinoculated intracerebrally (ic) with Rhabdoviridae is currently divided into nine approved ge- 15–20 μl, intraperitoneally (ip) with 100 μlorsubcutane- nera (Vesiculovirus, Perhavirus, Ephemerovirus, Lyssavirus, ously (sc) with 100 μlofastockofVero-grownFARV(CT Tibrovirus, Sigmavirus, Nucleorhabdovirus, Cytorhabdovirus AN 114) virus containing approximately 107 plaque and Novirhabdovirus);however,alargenumberofanimal forming units (PFU) per ml. -

And Γ- Cytoplasmic Actin in Vaccinia Virus Infection
Lights, Camera, Actin: Divergent roles of β- and γ- cytoplasmic actin in vaccinia virus infection NOORUL BISHARA MARZOOK A thesis submitted in fulfillment of requirements for the degree of Doctor of Philosophy FACULTY OF SCIENCE SCHOOL OF MOLECULAR BIOSCIENCE UNIVERSITY OF SYDNEY 2017 i TABLE OF CONTENTS Table of Contents ........................................................................................................... ii Acknowledgements ....................................................................................................... v Declaration ................................................................................................................... vii Abstract ....................................................................................................................... viii List of Figures ................................................................................................................ x List of Publications Arising From This Work.............................................................. xi Abbreviations Used ..................................................................................................... xii Chapter 1: Introduction ............................................................................................... 1 1.1 The Cytoskeleton ............................................................................................................ 2 1.1.1 The Eukaryotic Cytoskeleton ..................................................................................... -
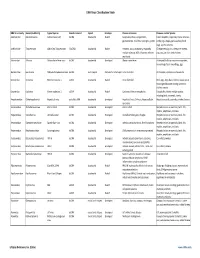
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae -

Viruses in Transplantation - Not Always Enemies
Viruses in transplantation - not always enemies Virome and transplantation ECCMID 2018 - Madrid Prof. Laurent Kaiser Head Division of Infectious Diseases Laboratory of Virology Geneva Center for Emerging Viral Diseases University Hospital of Geneva ESCMID eLibrary © by author Conflict of interest None ESCMID eLibrary © by author The human virome: definition? Repertoire of viruses found on the surface of/inside any body fluid/tissue • Eukaryotic DNA and RNA viruses • Prokaryotic DNA and RNA viruses (phages) 25 • The “main” viral community (up to 10 bacteriophages in humans) Haynes M. 2011, Metagenomic of the human body • Endogenous viral elements integrated into host chromosomes (8% of the human genome) • NGS is shaping the definition Rascovan N et al. Annu Rev Microbiol 2016;70:125-41 Popgeorgiev N et al. Intervirology 2013;56:395-412 Norman JM et al. Cell 2015;160:447-60 ESCMID eLibraryFoxman EF et al. Nat Rev Microbiol 2011;9:254-64 © by author Viruses routinely known to cause diseases (non exhaustive) Upper resp./oropharyngeal HSV 1 Influenza CNS Mumps virus Rhinovirus JC virus RSV Eye Herpes viruses Parainfluenza HSV Measles Coronavirus Adenovirus LCM virus Cytomegalovirus Flaviviruses Rabies HHV6 Poliovirus Heart Lower respiratory HTLV-1 Coxsackie B virus Rhinoviruses Parainfluenza virus HIV Coronaviruses Respiratory syncytial virus Parainfluenza virus Adenovirus Respiratory syncytial virus Coronaviruses Gastro-intestinal Influenza virus type A and B Human Bocavirus 1 Adenovirus Hepatitis virus type A, B, C, D, E Those that cause -

The Complete Genome of an Endogenous Nimavirus (Nimav-1 Lva) from the Pacific Whiteleg Shrimp Penaeus (Litopenaeus) Vannamei
G C A T T A C G G C A T genes Article The Complete Genome of an Endogenous Nimavirus (Nimav-1_LVa) From the Pacific Whiteleg Shrimp Penaeus (Litopenaeus) Vannamei Weidong Bao 1,* , Kathy F. J. Tang 2 and Acacia Alcivar-Warren 3,4,* 1 Genetic Information Research Institute, 20380 Town Center Lane, Suite 240, Cupertino, CA 95014, USA 2 Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, 106 Nanjing Road, Qingdao 266071, China; [email protected] 3 Fundación para la Conservation de la Biodiversidad Acuática y Terrestre (FUCOBI), Quito EC1701, Ecuador 4 Environmental Genomics Inc., ONE HEALTH Epigenomics Educational Initiative, P.O. Box 196, Southborough, MA 01772, USA * Correspondence: [email protected] (W.B.); [email protected] (A.A.-W.) Received: 17 December 2019; Accepted: 9 January 2020; Published: 14 January 2020 Abstract: White spot syndrome virus (WSSV), the lone virus of the genus Whispovirus under the family Nimaviridae, is one of the most devastating viruses affecting the shrimp farming industry. Knowledge about this virus, in particular, its evolution history, has been limited, partly due to its large genome and the lack of other closely related free-living viruses for comparative studies. In this study, we reconstructed a full-length endogenous nimavirus consensus genome, Nimav-1_LVa (279,905 bp), in the genome sequence of Penaeus (Litopenaeus) vannamei breed Kehai No. 1 (ASM378908v1). This endogenous virus seemed to insert exclusively into the telomeric pentanucleotide microsatellite (TAACC/GGTTA)n. It encoded 117 putative genes, with some containing introns, such as g012 (inhibitor of apoptosis, IAP), g046 (crustacean hyperglycemic hormone, CHH), g155 (innexin), g158 (Bax inhibitor 1 like). -

Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics
viruses Article Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics Ashleigh F. Porter 1, Mang Shi 1, John-Sebastian Eden 1,2 , Yong-Zhen Zhang 3,4 and Edward C. Holmes 1,3,* 1 Marie Bashir Institute for Infectious Diseases and Biosecurity, Charles Perkins Centre, School of Life & Environmental Sciences and Sydney Medical School, The University of Sydney, Sydney, NSW 2006, Australia; [email protected] (A.F.P.); [email protected] (M.S.); [email protected] (J.-S.E.) 2 Centre for Virus Research, Westmead Institute for Medical Research, Westmead, NSW 2145, Australia 3 Shanghai Public Health Clinical Center and School of Public Health, Fudan University, Shanghai 201500, China; [email protected] 4 Department of Zoonosis, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Changping, Beijing 102206, China * Correspondence: [email protected]; Tel.: +61-2-9351-5591 Received: 17 October 2019; Accepted: 23 November 2019; Published: 25 November 2019 Abstract: DNA viruses comprise a wide array of genome structures and infect diverse host species. To date, most studies of DNA viruses have focused on those with the strongest disease associations. Accordingly, there has been a marked lack of sampling of DNA viruses from invertebrates. Bulk RNA sequencing has resulted in the discovery of a myriad of novel RNA viruses, and herein we used this methodology to identify actively transcribing DNA viruses in meta-transcriptomic libraries of diverse invertebrate species. Our analysis revealed high levels of phylogenetic diversity in DNA viruses, including 13 species from the Parvoviridae, Circoviridae, and Genomoviridae families of single-stranded DNA virus families, and six double-stranded DNA virus species from the Nudiviridae, Polyomaviridae, and Herpesviridae, for which few invertebrate viruses have been identified to date. -

Diversity of Large DNA Viruses of Invertebrates ⇑ Trevor Williams A, Max Bergoin B, Monique M
Journal of Invertebrate Pathology 147 (2017) 4–22 Contents lists available at ScienceDirect Journal of Invertebrate Pathology journal homepage: www.elsevier.com/locate/jip Diversity of large DNA viruses of invertebrates ⇑ Trevor Williams a, Max Bergoin b, Monique M. van Oers c, a Instituto de Ecología AC, Xalapa, Veracruz 91070, Mexico b Laboratoire de Pathologie Comparée, Faculté des Sciences, Université Montpellier, Place Eugène Bataillon, 34095 Montpellier, France c Laboratory of Virology, Wageningen University, Droevendaalsesteeg 1, 6708 PB Wageningen, The Netherlands article info abstract Article history: In this review we provide an overview of the diversity of large DNA viruses known to be pathogenic for Received 22 June 2016 invertebrates. We present their taxonomical classification and describe the evolutionary relationships Revised 3 August 2016 among various groups of invertebrate-infecting viruses. We also indicate the relationships of the Accepted 4 August 2016 invertebrate viruses to viruses infecting mammals or other vertebrates. The shared characteristics of Available online 31 August 2016 the viruses within the various families are described, including the structure of the virus particle, genome properties, and gene expression strategies. Finally, we explain the transmission and mode of infection of Keywords: the most important viruses in these families and indicate, which orders of invertebrates are susceptible to Entomopoxvirus these pathogens. Iridovirus Ó Ascovirus 2016 Elsevier Inc. All rights reserved. Nudivirus Hytrosavirus Filamentous viruses of hymenopterans Mollusk-infecting herpesviruses 1. Introduction in the cytoplasm. This group comprises viruses in the families Poxviridae (subfamily Entomopoxvirinae) and Iridoviridae. The Invertebrate DNA viruses span several virus families, some of viruses in the family Ascoviridae are also discussed as part of which also include members that infect vertebrates, whereas other this group as their replication starts in the nucleus, which families are restricted to invertebrates. -

The Intestinal Virome of Malabsorption Syndrome-Affected and Unaffected
Virus Research 261 (2019) 9–20 Contents lists available at ScienceDirect Virus Research journal homepage: www.elsevier.com/locate/virusres The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics T ⁎ Diane A. Limaa, , Samuel P. Cibulskib, Caroline Tochettoa, Ana Paula M. Varelaa, Fabrine Finklera, Thais F. Teixeiraa, Márcia R. Loikoa, Cristine Cervac, Dennis M. Junqueirad, Fabiana Q. Mayerc, Paulo M. Roehea a Laboratório de Virologia, Departamento de Microbiologia, Imunologia e Parasitologia, Instituto de Ciências Básicas da Saúde (ICBS), Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brazil b Laboratório de Virologia, Faculdade de Veterinária, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil c Laboratório de Biologia Molecular, Instituto de Pesquisas Veterinárias Desidério Finamor (IPVDF), Eldorado do Sul, RS, Brazil d Centro Universitário Ritter dos Reis - UniRitter, Health Science Department, Porto Alegre, RS, Brazil ARTICLE INFO ABSTRACT Keywords: Malabsorption syndrome (MAS) is an economically important disease of young, commercially reared broilers, Enteric disorders characterized by growth retardation, defective feather development and diarrheic faeces. Several viruses have Virome been tentatively associated to such syndrome. Here, in order to examine potential associations between enteric Broiler chickens viruses and MAS, the faecal viromes of 70 stool samples collected from diseased (n = 35) and healthy (n = 35) High-throughput sequencing chickens from seven flocks were characterized and compared. Following high-throughput sequencing, a total of 8,347,319 paired end reads, with an average of 231 nt, were generated. Through analysis of de novo assembled contigs, 144 contigs > 1000 nt were identified with hits to eukaryotic viral sequences, as determined by GenBank database.