Phosphorylating Titin's Cardiac N2B Element by ERK2 Or Camkii&Delta
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Regulation of Titin-Based Cardiac Stiffness by Unfolded Domain Oxidation (Undox)
Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx) Christine M. Loeschera,1, Martin Breitkreuzb,1, Yong Lia, Alexander Nickelc, Andreas Ungera, Alexander Dietld, Andreas Schmidte, Belal A. Mohamedf, Sebastian Kötterg, Joachim P. Schmitth, Marcus Krügere,i, Martina Krügerg, Karl Toischerf, Christoph Maackc, Lars I. Leichertj, Nazha Hamdanib, and Wolfgang A. Linkea,2 aInstitute of Physiology II, University of Munster, 48149 Munster, Germany; bInstitute of Physiology, Ruhr University Bochum, 44801 Bochum, Germany; cComprehensive Heart Failure Center Wuerzburg, University Clinic Wuerzburg, 97078 Wuerzburg, Germany; dDepartment of Internal Medicine II, University Hospital Regensburg, 93053 Regensburg, Germany; eInstitute for Genetics, University of Cologne, 50931 Cologne, Germany; fDepartment of Cardiology and Pneumology, University Medicine Goettingen, 37075 Goettingen, Germany; gDepartment of Cardiovascular Physiology, Heinrich Heine University, 40225 Düsseldorf, Germany; hDepartment of Pharmacology and Clinical Pharmacology, Heinrich Heine University, 40225 Düsseldorf, Germany; iCenter for Molecular Medicine and Excellence Cluster "Cellular Stress Responses in Aging-Associated Diseases" (CECAD), University of Cologne, 50931 Cologne, Germany; and jInstitute for Biochemistry and Pathobiochemistry, Ruhr University Bochum, 44801 Bochum, Germany Edited by Jonathan Seidman, Harvard University, Boston, MA, and approved August 12, 2020 (received for review March 14, 2020) The relationship between oxidative stress and -

Postmortem Changes in the Myofibrillar and Other Cytoskeletal Proteins in Muscle
BIOCHEMISTRY - IMPACT ON MEAT TENDERNESS Postmortem Changes in the Myofibrillar and Other C'oskeletal Proteins in Muscle RICHARD M. ROBSON*, ELISABETH HUFF-LONERGAN', FREDERICK C. PARRISH, JR., CHIUNG-YING HO, MARVIN H. STROMER, TED W. HUIATT, ROBERT M. BELLIN and SUZANNE W. SERNETT introduction filaments (titin), and integral Z-line region (a-actinin, Cap Z), as well as proteins of the intermediate filaments (desmin, The cytoskeleton of "typical" vertebrate cells contains paranemin, and synemin), Z-line periphery (filamin) and three protein filament systems, namely the -7-nm diameter costameres underlying the cell membrane (filamin, actin-containing microfilaments, the -1 0-nm diameter in- dystrophin, talin, and vinculin) are listed along with an esti- termediate filaments (IFs), and the -23-nm diameter tubu- mate of their abundance, approximate molecular weights, lin-containing microtubules (Robson, 1989, 1995; Robson and number of subunits per molecule. Because the myofibrils et al., 1991 ).The contractile myofibrils, which are by far the are the overwhelming components of the skeletal muscle cell major components of developed skeletal muscle cells and cytoskeleton, the approximate percentages of the cytoskel- are responsible for most of the desirable qualities of muscle eton listed for the myofibrillar proteins (e.g., myosin, actin, foods (Robson et al., 1981,1984, 1991 1, can be considered tropomyosin, a-actinin, etc.) also would represent their ap- the highly expanded corollary of the microfilament system proximate percentages of total myofibrillar protein. of non-muscle cells. The myofibrils, IFs, cell membrane skel- eton (complex protein-lattice subjacent to the sarcolemma), Some Important Characteristics, Possible and attachment sites connecting these elements will be con- Roles, and Postmortem Changes of Key sidered as comprising the muscle cell cytoskeleton in this Cytoskeletal Proteins review. -

Α-Actinin/Titin Interaction: a Dynamic and Mechanically Stable Cluster of Bonds in the Muscle Z-Disk
α-Actinin/titin interaction: A dynamic and mechanically stable cluster of bonds in the muscle Z-disk Marco Grisona, Ulrich Merkela, Julius Kostanb, Kristina Djinovic-Carugob,c, and Matthias Riefa,d,1 aPhysik Department E22, Technische Universität München, 85748 Garching, Germany; bDepartment of Structural and Computational Biology, Max F. Perutz Laboratories, University of Vienna, A-1030 Vienna, Austria; cDepartment of Biochemistry, Faculty of Chemistry and Chemical Technology, University of Ljubljana, SI-1000 Ljubljana, Slovenia; and dMunich Center for Integrated Protein Science, 81377 Munich, Germany Edited by James A. Spudich, Stanford University School of Medicine, Stanford, CA, and approved December 16, 2016 (received for review August 2, 2016) Stable anchoring of titin within the muscle Z-disk is essential for In humans, the isoforms of titin exhibit four to seven Z-repeats preserving muscle integrity during passive stretching. One of the (15, 16, 20). The structure of the EF3-4 hands complex with titin main candidates for anchoring titin in the Z-disk is the actin cross- Z-repeat 7 shows the bound Z-repeat in an α-helical confor- linker α-actinin. The calmodulin-like domain of α-actinin binds to mation (21). In solution assays, binding affinities of various the Z-repeats of titin. However, the mechanical and kinetic prop- Z-repeats to EF3-4 were determined to lie in the micromolar erties of this important interaction are still unknown. Here, we use range (22). Micromolar affinity points only to a moderately a dual-beam optical tweezers assay to study the mechanics of this stable interaction, the kinetics of which are unknown. -
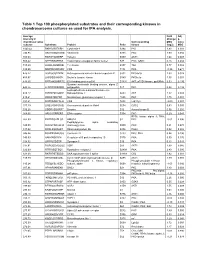
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Association of Titin and Myosin Heavy Chain in Developing Skeletal Muscle (Myogenesis/Cytoskeleton/Assembly in Vvo) W
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 74%-7500, August 1992 Cell Biology Association of titin and myosin heavy chain in developing skeletal muscle (myogenesis/cytoskeleton/assembly in vvo) W. B. ISAACS*, I. S. KIM, A. STRUVE, AND A. B. FULTONt Department of Biochemistry, University of Iowa, Iowa City, IA 52242 Communicated by Sheldon Penman, May 22, 1992 ABSTRACT To understand molecular interactions that deficient medium (10). After labeling, cultures either were organize developing myoflbrils, we examined the biosynthesis extracted immediately or were chased by adding complete and interaction of titin and myosin heavy chain in cultures of medium supplemented with 2 mM unlabeled methionine and developing muscle. Use of pulse-labeling, immunoprecipita- incubating at 370C for various times before extraction. Ex- tion, and a reversible cross-linking procedure demonstrates tractions used 0.5% Triton X-100 in extraction buffer (100 that within minutes of synthesis, titin and myosin heavy chain mM KCI/10 mM Pipes, pH 6.8/300 mM sucrose/2 mM can be chemically cross-linked into very large, detergent- MgCI2/1 mM EGTA) containing protease inhibitors (1 mM resistant complexes retaining many features of intact myo- phenylmethylsulfonyl fluoride and 100 mM leupeptin; ref. tubes. These complexes, predominantly of titin and myosin, 11). occur very early in myofibrillogenesis as well as later. These Immunoprecipitation. Titin was precipitated by using a data suggest that synthesis and assembly oftitin and myosin are mouse monoclonal antibody (mAb), AMF-1, as described temporally and spatially coordinated in nascent myofibrils and (10). Muscle-specific myosin heavy chain (hereafter myosin) support the hypothesis that titin molecules help to organize was precipitated with mAb MF-20 (12), a gift ofD. -

Titin N2A Domain and Its Interactions at the Sarcomere
International Journal of Molecular Sciences Review Titin N2A Domain and Its Interactions at the Sarcomere Adeleye O. Adewale and Young-Hoon Ahn * Department of Chemistry, Wayne State University, Detroit, MI 48202, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-(313)-577-1384 Abstract: Titin is a giant protein in the sarcomere that plays an essential role in muscle contraction with actin and myosin filaments. However, its utility goes beyond mechanical functions, extending to versatile and complex roles in sarcomere organization and maintenance, passive force, mechanosens- ing, and signaling. Titin’s multiple functions are in part attributed to its large size and modular structures that interact with a myriad of protein partners. Among titin’s domains, the N2A element is one of titin’s unique segments that contributes to titin’s functions in compliance, contraction, structural stability, and signaling via protein–protein interactions with actin filament, chaperones, stress-sensing proteins, and proteases. Considering the significance of N2A, this review highlights structural conformations of N2A, its predisposition for protein–protein interactions, and its multiple interacting protein partners that allow the modulation of titin’s biological effects. Lastly, the nature of N2A for interactions with chaperones and proteases is included, presenting it as an important node that impacts titin’s structural and functional integrity. Keywords: titin; N2A domain; protein–protein interaction 1. Introduction Citation: Adewale, A.O.; Ahn, Y.-H. The complexity of striated muscle is defined by the intricate organization of its com- Titin N2A Domain and Its ponents [1]. The involuntary cardiac and voluntary skeletal muscles are the primary types Interactions at the Sarcomere. -

B3GALNT2 Is a Gene Associated with Congenital Muscular Dystrophy with Brain Malformations
European Journal of Human Genetics (2014) 22, 707–710 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg SHORT REPORT B3GALNT2 is a gene associated with congenital muscular dystrophy with brain malformations Carola Hedberg*,1, Anders Oldfors1 and Niklas Darin2 Congenital muscular dystrophies associated with brain malformations are a group of disorders frequently associated with aberrant glycosylation of a-dystroglycan. They include disease entities such a Walker–Warburg syndrome, muscle–eye–brain disease and various other clinical phenotypes. Different genes involved in glycosylation of a-dystroglycan are associated with these dystroglycanopathies. We describe a 5-year-old girl with psychomotor retardation, ataxia, spasticity, muscle weakness and increased serum creatine kinase levels. Immunhistochemistry of skeletal muscle revealed reduced glycosylated a-dystroglycan. Magnetic resonance imaging of the brain at 3.5 years of age showed increased T2 signal from supratentorial and infratentorial white matter, a hypoplastic pons and subcortical cerebellar cysts. By whole exome sequencing, the patient was identified to be compound heterozygous for a one-base duplication and a missense mutation in the gene B3GALNT2 (b-1,3-N-acetylgalactos- aminyltransferase 2; B3GalNAc-T2). This patient showed a milder phenotype than previously described patients with mutations in the B3GALNT2 gene. European Journal of Human Genetics (2014) 22, 707–710; doi:10.1038/ejhg.2013.223; published online 2 October 2013 Keywords: -

Identification of Cyclin-Dependent Kinase 5 in T Cells and Its
IDENTIFICATION OF CYCLIN-DEPENDENT KINASE 5 IN T CELLS AND ITS ROLE IN REGULATING T CELL FUNCTION AND DIFFERENTIATION By ERIC M. LAM Submitted in partial fulfillment of the requirements For the degree of Doctor of Philosophy Dissertation Advisor: Dr. John J. Letterio, MD Department of Pharmacology CASE WESTERN RESERVE UNIVERSITY May, 2015 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of ERIC M LAM candidate for the degree of Doctor of Philosophy degree *. Committee Members Noa Noy (Committee Chair) John J Letterio (Mentor) Bing-Cheng Wang Anthony Berdis Marvin Nieman Date of Defense January 13, 2015 *We also certify that written approval has been obtained for any proprietary material contained therein. Dedication To my parents, who scarified everything for their children to have a better life, and taught us to work hard, work smart, and to love your family and friends. To my sister who has always inspired me and taken care of me. Thank you. Table of Contents Table of Contents i List of Figures/Tables iv Acknowledgements vi Abstract 1 Chapter 1: Introduction 3 1.1 Overview of Cyclin-dependent kinase 5 (Cdk5) 4 1.1.1 History of Cdk5 discovery and introduction to an atypical CDK 4 1.1.2 Regulation of Cdk5 activity 5 1.1.3 Function of Cdk5 in the nervous system 6 1.1.4 Dysregulation of Cdk5 in disease and disorder 9 1.1.5 Cdk5 and its role in non-neuronal tissues 10 1.2 The Immune System and Regulatory T cells 13 1.2.1 T cell subsets 14 1.2.2 The role of IL2 in T cell function and the -

Actin-Titin Interaction in Cardiac Myofibrils: Probing a Physiological Role
Biophysical Journal Volume 73 August 1997 905-919 905 Actin-Titin Interaction in Cardiac Myofibrils: Probing a Physiological Role Wolfgang A. Linke,* Marc lvemeyer,* Siegfried Labeit,# Horst Hinssen,§ J. Caspar Ruegg,* and Mathias Gautel# *Institute of Physiology II, University of Heidelberg, D-69120 Heidelberg; #European Molecular Biology Laboratory, D-69012 Heidelberg; and §Biochemical Cell Biology Group, University of Bielefeld, D-33501 Bielefeld, Germany ABSTRACT The high stiffness of relaxed cardiac myofibrils is explainable mainly by the expression of a short-length titin (connectin), the giant elastic protein of the vertebrate myofibrillar cytoskeleton. However, additional molecular features could account for this high stiffness, such as interaction between titin and actin, which has previously been reported in vitro. To probe this finding for a possible physiological significance, isolated myofibrils from rat heart were subjected to selective removal of actin filaments by a calcium-independent gelsolin fragment, and the "passive" stiffness of the specimens was recorded. Upon actin extraction, stiffness decreased by nearly 60%, and to a similar degree after high-salt extraction of thick filaments. Thus actin-titin association indeed contributes to the stiffness of resting cardiac muscle. To identify possible sites of association, we employed a combination of different techniques. Immunofluorescence microscopy revealed that actin extraction increased the extensibility of the previously stiff Z-disc-flanking titin region. Actin-titin interaction within this region was confirmed in in vitro cosedimentation assays, in which multimodule recombinant titin fragments were tested for their ability to interact with F-actin. By contrast, such assays showed no actin-titin-binding propensity for sarcomeric regions outside the Z-disc comb. -

Elucidating the Unknown Role of Cyclin Dependent Kinase 5 in Cardiac Pathophysiological
Elucidating the Unknown Role of Cyclin Dependent Kinase 5 in Cardiac Pathophysiological Conditions Danielle Aina-Badejo Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2021 © 2021 Danielle Aina-Badejo All Rights Reserved Abstract Elucidating the Unknown Role of Cyclin Dependent Kinase 5 in Cardiac Pathophysiological Conditions Danielle Aina-Badejo Until now, the role of cyclin dependent kinase 5 (CDK5) in cardiac pathophysiology has not been explored. While CDK5 has been well studied in the neuroscience/Alzheimer’s field as a cyclin-independent kinase, there is currently no investigation into the cardiac-specific role of CDK5. Recently, it was established that inhibition of CDK5 in stem cell derived cardiomyocytes from individuals with Timothy Syndrome (TS) rescued the delayed inactivation phenotype; TS is a fatal genetic long QT syndrome (LQTS) caused by delayed inactivation of the L-type voltage 2+ gated Ca channel Ca V1.2. While it is evident that CDK5 plays an important role in regulating Ca V1.2 function, its role in cardiac tissue remains to be elucidated. To determine whether CDK5 is essential for cardiac function, two separate mouse models were established—a cardiac-deficient Cdk5 mouse model ( Cdk5 flox x αMHC-MerCreMer +) and a Cdk5 activation mouse model via overexpression of Cdk5’s known activator, p35 (Cdk5r1/ p35 OE x αMHC-MerCreMer +). Immediately after spatiotemporal induction of deficiency/activation of Cdk5 in adult mice, echocardiography, histology and proteomic analysis were performed to examine effects on cardiac structure and function. -

Muscle Diseases: the Muscular Dystrophies
ANRV295-PM02-04 ARI 13 December 2006 2:57 Muscle Diseases: The Muscular Dystrophies Elizabeth M. McNally and Peter Pytel Department of Medicine, Section of Cardiology, University of Chicago, Chicago, Illinois 60637; email: [email protected] Department of Pathology, University of Chicago, Chicago, Illinois 60637; email: [email protected] Annu. Rev. Pathol. Mech. Dis. 2007. Key Words 2:87–109 myotonia, sarcopenia, muscle regeneration, dystrophin, lamin A/C, The Annual Review of Pathology: Mechanisms of Disease is online at nucleotide repeat expansion pathmechdis.annualreviews.org Abstract by Drexel University on 01/13/13. For personal use only. This article’s doi: 10.1146/annurev.pathol.2.010506.091936 Dystrophic muscle disease can occur at any age. Early- or childhood- onset muscular dystrophies may be associated with profound loss Copyright c 2007 by Annual Reviews. All rights reserved of muscle function, affecting ambulation, posture, and cardiac and respiratory function. Late-onset muscular dystrophies or myopathies 1553-4006/07/0228-0087$20.00 Annu. Rev. Pathol. Mech. Dis. 2007.2:87-109. Downloaded from www.annualreviews.org may be mild and associated with slight weakness and an inability to increase muscle mass. The phenotype of muscular dystrophy is an endpoint that arises from a diverse set of genetic pathways. Genes associated with muscular dystrophies encode proteins of the plasma membrane and extracellular matrix, and the sarcomere and Z band, as well as nuclear membrane components. Because muscle has such distinctive structural and regenerative properties, many of the genes implicated in these disorders target pathways unique to muscle or more highly expressed in muscle. -

Molecular Interactions Among the Intermediate Filament Proteins Paranemin, Synemin and Desmin and Their Localization During Skeletal Muscle Cell Development
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1-1-2002 Molecular interactions among the intermediate filament proteins paranemin, synemin and desmin and their localization during skeletal muscle cell development Stephanie Ann Lex Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Recommended Citation Lex, Stephanie Ann, "Molecular interactions among the intermediate filament proteins paranemin, synemin and desmin and their localization during skeletal muscle cell development" (2002). Retrospective Theses and Dissertations. 20141. https://lib.dr.iastate.edu/rtd/20141 This Thesis is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Molecular interactions among the intermediate filament proteins paranemin, synemin and desmin and their localization during skeletal muscle cell development by Stephanie Ann Lex A thesis submitted to the graduate faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Major: Biochemistry Program of Study Committee: Richard M. Robson, Major Professor Elizabeth J. Huff-Lonergan Ted W. Huiatt Marvin H. Stromer Iowa State University Ames, Iowa 2002 11 Graduate College Iowa State University