SAR of Benzodiazepines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -
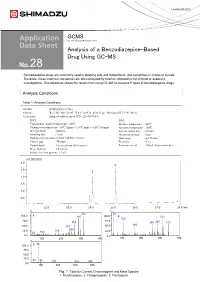
Analysis of a Benzodiazepine-Based Drug Using GC-MS 28
LAAN-E-MS-E028 GCMS Gas Chromatograph Mass Spectrometer Analysis of a Benzodiazepine-Based Drug Using GC-MS 28 Benzodiazepine drugs are commonly used in sleeping aids and tranquilizers, and sometimes in crimes or suicide. Therefore, these chemical substances are often analyzed by forensic laboratories for criminal or academic investigations. This datasheet shows the results from using GC-MS to measure 9 types of benzodiazepine drugs. Analysis Conditions Table 1: Analysis Conditions GC-MS :GCMS-QP2010 Ultra Column : Rxi®-5Sil MS (30 mL. X 0.25 mmI.D., df=0.25 µm, Shimadzu GLC P/N:13623) Glass insert :Silanized splitless insert (P/N: 221-48876-03) [GC] [MS] Vaporization chamber temperature : 260℃ Interface temperature : 280℃ Column oven temperature : 60℃ (2min) -> (10℃/min) -> 320℃ (10min) Ion source temperature : 200℃ Injection mode : Splitless Solvent elution time : 2.0 min Sampling time : 1 min Measurement mode : Scan High pressure injection method: 250 kPa (1.5 min) Mass range : m/z 35-600 Carrier gas : Helium Event time : 0.3 sec Control mode :Linear velocity (45.6 cm/sec) Emission current : 150 µA (high sensitivity) Purge flow rate :3.0 ml/min Sample injection quantity :1.0 µL (x1,000,000) 3.0 3 2.5 2 2.0 1.5 1.0 1 0.5 22.0 23.0 24.0 25.0 26.0 27.0 28.0 min % % 312 55 100.0 1 285 100.0 2 313 109 75.0 75.0 266 259287 342 166 50.0 238 50.0 248 25.0 183 25.0 63 109 0.0 0.0 100 200 300 400 100 200 300 400 % 86 100.0 3 75.0 50.0 25.0 58 99 183 315 387 0.0 100 200 300 400 Fig. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0005722 A1 Jennings-Spring (43) Pub
US 20090005722A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0005722 A1 Jennings-Spring (43) Pub. Date: Jan. 1, 2009 (54) SKIN-CONTACTING-ADHESIVE FREE Publication Classification DRESSING (51) Int. Cl. Inventor: Barbara Jennings-Spring, Jupiter, A61N L/30 (2006.01) (76) A6F I3/00 (2006.01) FL (US) A6IL I5/00 (2006.01) Correspondence Address: AOIG 7/06 (2006.01) Irving M. Fishman AOIG 7/04 (2006.01) c/o Cohen, Tauber, Spievack and Wagner (52) U.S. Cl. .................. 604/20: 602/43: 602/48; 4771.5; Suite 2400, 420 Lexington Avenue 47/13 New York, NY 10170 (US) (57) ABSTRACT (21) Appl. No.: 12/231,104 A dressing having a flexible sleeve shaped to accommodate a Substantially cylindrical body portion, the sleeve having a (22) Filed: Aug. 29, 2008 lining which is substantially non-adherent to the body part being bandaged and having a peripheral securement means Related U.S. Application Data which attaches two peripheral portions to each other without (63) Continuation-in-part of application No. 1 1/434,689, those portions being circumferentially adhered to the sleeve filed on May 16, 2006. portion. Patent Application Publication Jan. 1, 2009 Sheet 1 of 9 US 2009/0005722 A1 Patent Application Publication Jan. 1, 2009 Sheet 2 of 9 US 2009/0005722 A1 10 8 F.G. 5 Patent Application Publication Jan. 1, 2009 Sheet 3 of 9 US 2009/0005722 A1 13 FIG.6 2 - Y TIII Till "T fift 11 10 FIG.7 8 13 6 - 12 - Timir" "in "in "MINIII. -

Drug and Medication Classification Schedule
KENTUCKY HORSE RACING COMMISSION UNIFORM DRUG, MEDICATION, AND SUBSTANCE CLASSIFICATION SCHEDULE KHRC 8-020-1 (11/2018) Class A drugs, medications, and substances are those (1) that have the highest potential to influence performance in the equine athlete, regardless of their approval by the United States Food and Drug Administration, or (2) that lack approval by the United States Food and Drug Administration but have pharmacologic effects similar to certain Class B drugs, medications, or substances that are approved by the United States Food and Drug Administration. Acecarbromal Bolasterone Cimaterol Divalproex Fluanisone Acetophenazine Boldione Citalopram Dixyrazine Fludiazepam Adinazolam Brimondine Cllibucaine Donepezil Flunitrazepam Alcuronium Bromazepam Clobazam Dopamine Fluopromazine Alfentanil Bromfenac Clocapramine Doxacurium Fluoresone Almotriptan Bromisovalum Clomethiazole Doxapram Fluoxetine Alphaprodine Bromocriptine Clomipramine Doxazosin Flupenthixol Alpidem Bromperidol Clonazepam Doxefazepam Flupirtine Alprazolam Brotizolam Clorazepate Doxepin Flurazepam Alprenolol Bufexamac Clormecaine Droperidol Fluspirilene Althesin Bupivacaine Clostebol Duloxetine Flutoprazepam Aminorex Buprenorphine Clothiapine Eletriptan Fluvoxamine Amisulpride Buspirone Clotiazepam Enalapril Formebolone Amitriptyline Bupropion Cloxazolam Enciprazine Fosinopril Amobarbital Butabartital Clozapine Endorphins Furzabol Amoxapine Butacaine Cobratoxin Enkephalins Galantamine Amperozide Butalbital Cocaine Ephedrine Gallamine Amphetamine Butanilicaine Codeine -

II.3.4 Benzodiazepines by Hiroshi Seno and Hideki Hattori
3.4 II.3.4 Benzodiazepines by Hiroshi Seno and Hideki Hattori Introduction Benzodiazepines show antianxiety, hypnotic, anticonvulsant and muscle-relaxant eff ects. Th is group of drugs has wide safety dose ranges; it means that the ratio of the LD50 to the ED50 (therapeutic index) is high. Because of its safety, benzodiazepines are being widely used in the world. Some of benzodiazepines are also being abused or used for so-called “ drug facilitated sexual assault”, and thus they are under the control of the Narcotics and Psychotropics Control Law; in Japan, triazolam abuse has become one of the serious social problems. In this chapter, a GC/MS method for simultaneous analysis of 22 kinds of benzodiazepines listed in > Table 4.1 is described. In addition, the LC/MS analysis of triazolam, and its metabolites 4-hydroxy- triazolam and α-hydroxytriazolam is also presented. GC/MS analysis of benzodiazepines in blood and urine Reagents and their preparation • Th e pure powder of the 22 kinds of benzodiazepines was donated by each pharmaceutical manufacturers according to the authors’ request a (some of benzodiazepines now obtaina- ble from Sigma, St. Louis, MO, USA). • 1 M Sodium bicarbonate solution: a 8.4-g aliquot of sodium bicarbonate is dissolved in distilled water to prepare 100 mL solution. • 2 M Sodium acetate solution: a 27.5-g aliquot of sodium acetate is dissolved in distilled water to prepare 100 mL solution. GC/MS conditions Column: a DB-5 fused silica capillary column (30 m × 0.25 mm i.d., fi lm thickness 0.25 µm, J & W Scientifi c, Folsom, CA, USA). -

Muscle Relaxants for Pain Management in Rheumatoid Arthritis (Review)
Muscle relaxants for pain management in rheumatoid arthritis (Review) Richards BL, Whittle SL, Buchbinder R This is a reprint of a Cochrane review, prepared and maintained by The Cochrane Collaboration and published in The Cochrane Library 2012, Issue 1 http://www.thecochranelibrary.com Muscle relaxants for pain management in rheumatoid arthritis (Review) Copyright © 2012 The Cochrane Collaboration. Published by John Wiley & Sons, Ltd. TABLE OF CONTENTS HEADER....................................... 1 ABSTRACT ...................................... 1 PLAINLANGUAGESUMMARY . 2 SUMMARY OF FINDINGS FOR THE MAIN COMPARISON . ..... 3 BACKGROUND .................................... 6 OBJECTIVES ..................................... 7 METHODS ...................................... 7 RESULTS....................................... 10 Figure1. ..................................... 11 Figure2. ..................................... 13 Figure3. ..................................... 15 Figure4. ..................................... 15 Figure5. ..................................... 16 Figure6. ..................................... 17 Figure7. ..................................... 17 Figure8. ..................................... 18 DISCUSSION ..................................... 20 AUTHORS’CONCLUSIONS . 21 ACKNOWLEDGEMENTS . 22 REFERENCES ..................................... 22 CHARACTERISTICSOFSTUDIES . 24 DATAANDANALYSES. 35 Analysis 1.1. Comparison 1 Muscle relaxant versus control, Outcome 1 Pain 24hrs. 37 Analysis 1.2. Comparison 1 Muscle relaxant -

Study on Raffenetti's P File Format in Conventional Ab Initio Self-Consistent-Field Molecular Orbital Calculations in Parallel
J. Comput. Chem. Jpn., Vol. 7, No. 5, pp. 179–184 (2008) c Society of Computer Chemistry, Japan Study on Raffenetti’s P File Format in Conventional Ab Initio Self-Consistent-Field Molecular Orbital Calculations in Parallel Computational Environment Hiroyuki TERAMAEa* and Kazushige OHTAWARAb,c aDepartment of Chemistry, Faculty of Science, Josai University 1-1 Keyakidai, Sakado, Saitama 350-0295, Japan bATR Adaptive Communication Research Laboratories 2-2-2 Hikaridai, Seika-cho, Soraku-gun, Kyoto 619-0288, Japan c Present address: Technology Development Division, Victor Company of Japan, Ltd. 58-7 Shinmei-cho, Yokosuka, Kanagawa 239-8550, Japan *e-mail: [email protected] (Received: August 7, 2008; Accepted for publication: October 17, 2008; Advance publication: November 22, 2008) We compare the CPU time and the wall clock time of the Raffenetti’s P file algorithm with the usual algorithm on the two electron integrals storing with four suffixes of the ab initio Hartree-Fock calculations. The calculations are performed with the flutoprazepam, triazolam, clotiazepam, etizolam, and flutazolam molecules. These molecules are all minor-tranquilizers with the benzodiazepine or thienodiazepine backbone. The 3-21G basis sets are employed. Almost in all cases, P file algorithm gave slower speed than the usual algorithm. The number of two electron integrals increases almost two times larger than the usual algorithms. In a large molecule, the matrix of the two electron integrals becomes very sparse and the recombination of the integrals just increases the total number of the integrals. It is concluded that the P method sometimes calculates faster but sometimes does not. In a large scale calculation, it should be suggested to perform a test calculation to confirm which method is faster prior to the real calculations. -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -

PMDA Alert for Proper Use of Drugs When Using Benzodiazepine
■ PMDA Alert for Proper Use of Drugs https://www.pmda.go.jp/english/safety/info-services/drugs/properly- No. 11 March 2017 use-alert/0001.html PMDA Alert for Proper Use of Drugs Pharmaceuticals and Medical Devices Agency No. 11 March 2017 Dependence associated with Benzodiazepine Receptor Agonists [To Patients] This document is for healthcare professionals. If taking the drug, please consult with your physicians or pharmacists. Please don’t reduce the dosage or stop taking the drug on self-judgment. Benzodiazepine receptor agonists have a characteristic of developing physical dependence with long-term use even within an approved dose range, leading to various withdrawal symptoms on dose reduction or discontinuation. <Major withdrawal symptoms> insomnia, anxiety, feeling irritated, headache, queasy/vomiting, delirium, tremor, seizure, etc. Please pay careful attention to the following when using benzodiazepine receptor agonists as hypnotics-sedatives and anxiolytics. Healthcare professionals should avoid long-term use with chronic administration. - Dependence may occur with long-term use even within an approved dose range. - Therapeutic necessity should be carefully considered when continuing administration of the drug. Healthcare professionals should adhere to the dosage and confirm that there is no multiple prescription of similar drugs. - Long-term administration, high-dose administration, or multiple medications increase the risk of developing dependence. - Healthcare professionals should confirm that similar drugs are not prescribed by other medical institutions. Healthcare professionals should reduce the dose or discontinue carefully such as by gradual dose reduction or alternate-days administration when discontinuing the administration. - Sudden discontinuation will develop serious withdrawal symptoms in addition to aggravate primary diseases. -

State of Knowledge of Drug-Impaired Driving
State of Knowledge of Drug-Impaired Driving FINAL REPORT Technical Report Documentation Page 1. Report No. 2. Government Accession No. 3. Recipient's Catalog No. DOT HS 809 642 4. Title and Subtitle 5. Report Date September 2003 State of Knowledge of Drug-Impaired Driving 6. Performing Organization Code 7. Author(s) 8. Performing Organization Report No. Jones, R. K.; Shinar, D.; and Walsh, J. M. 9. Performing Organization Name and Address 10. Work Unit No. (TRAIS) Mid-America Research Institute 611 Main Street Winchester, MA 01890 11. Contract or Grant No. DTNH22-98-D-25079 12. Sponsoring Agency Name and Address 13. Type of Report and Period Covered National Highway Traffic Safety Administration Final Report Office of Research and Technology 400 7th Street, S.W. 14. Sponsoring Agency Code Washington, DC 20590 15. Supplementary Notes Amy Berning and Richard Compton were the Contracting Officer’s Technical Representatives for this project. 16. Abstract Examines the current state of knowledge of drug-impaired driving. The review covers a broad range of related research, including the detection and measurement of drugs in drivers, experimental research on the effect of drugs on the performance driving-related tasks, drug prevalence in various populations of drivers, drug-crash risk, and countermeasures for drug-impaired driving. The review covers scientific literature published since 1980. 17. Key Words 18. Distribution Statement drug-impaired drivers, experimental research, This report is available from the National Technical drug detection, drug measurement, Information Service, Springfield, Virginia 22161, epidemiologic research, drug-related traffic (703) 605-6000, and free of charge from the crashes, driving under the influence of drugs, NHTSA web site at www.nhtsa.dot.gov drug-crash countermeasures 19. -

TEXAS RACING COMMISSION January 28, 2021 To
TEXAS RACING COMMISSION P. O. Box 12080, Austin, Texas 78711-2080 8505 Cross Park Drive, Suite 110, Austin, Texas 78754-4552 Phone (512) 833-6699 Fax (512) 833-6907 www.txrc.texas.gov January 28, 2021 To: Stewards, Commission Veterinarians, Test Barn Supervisors, Practicing Veterinarians, Owners, and Trainers From: Chuck Trout, Executive Director Re: Effective February 25, 2021 changes to the following documents: • Permissible Levels of Therapeutic Medications and Naturally Occurring Substances • Equine Medication Classification Policy and Penalty Guidelines • Equine Medication Classification List. This memo is to provide notice that the above listed documents are to be replaced effective this date. The changes include, but are not limited to: • Changes to the list of Permissible Levels of Therapeutic Medications and Naturally Occurring Substances; • Changes to the Equine Medication Classification Policy and Penalty Guidelines; • Changes to the Equine Medication Classification List. These documents are subject to further revision at any time. Test Barn Supervisors - please post this memo and the revised documents in the test barn as soon as possible. Also, please distribute copies of the Permissible Levels of Therapeutic Medications and Naturally Occurring Substances and Equine Medication Classification List to the practicing veterinarians at your racetrack. Licensing Staff - please post this memo and the revised documents where they may be viewed by the public as soon as possible. Copies of these documents will be made available on the Commission's website at http://www.txrc.texas.gov. Attachments: Permissible Levels of Therapeutic Medications and Naturally Occurring Substances Equine Medication Classification Policy and Penalty Guidelines Equine Medication Classification List TEXAS RACING COMMISSION P. -

2021 Equine Prohibited Substances List
2021 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). Prohibited Substances that are identified as Specified Substances in the List below should not in any way be considered less important or less dangerous than other Prohibited Substances. Rather, they are simply substances which are more likely to have been ingested by Horses for a purpose other than the enhancement of sport performance, for example, through a contaminated food substance. LISTED AS SUBSTANCE ACTIVITY BANNED 1-androsterone Anabolic BANNED 3β-Hydroxy-5α-androstan-17-one Anabolic BANNED 4-chlorometatandienone Anabolic BANNED 5α-Androst-2-ene-17one Anabolic BANNED 5α-Androstane-3α, 17α-diol Anabolic BANNED 5α-Androstane-3α, 17β-diol Anabolic BANNED 5α-Androstane-3β, 17α-diol Anabolic BANNED 5α-Androstane-3β, 17β-diol Anabolic BANNED 5β-Androstane-3α, 17β-diol Anabolic BANNED 7α-Hydroxy-DHEA Anabolic BANNED 7β-Hydroxy-DHEA Anabolic BANNED 7-Keto-DHEA Anabolic CONTROLLED 17-Alpha-Hydroxy Progesterone Hormone FEMALES BANNED 17-Alpha-Hydroxy Progesterone Anabolic MALES BANNED 19-Norandrosterone Anabolic BANNED 19-Noretiocholanolone Anabolic BANNED 20-Hydroxyecdysone Anabolic BANNED Δ1-Testosterone Anabolic BANNED Acebutolol Beta blocker BANNED Acefylline Bronchodilator BANNED Acemetacin Non-steroidal anti-inflammatory drug BANNED Acenocoumarol Anticoagulant CONTROLLED Acepromazine Sedative BANNED Acetanilid Analgesic/antipyretic CONTROLLED Acetazolamide Carbonic Anhydrase Inhibitor BANNED Acetohexamide Pancreatic stimulant CONTROLLED Acetominophen (Paracetamol) Analgesic BANNED Acetophenazine Antipsychotic BANNED Acetophenetidin (Phenacetin) Analgesic BANNED Acetylmorphine Narcotic BANNED Adinazolam Anxiolytic BANNED Adiphenine Antispasmodic BANNED Adrafinil Stimulant 1 December 2020, Lausanne, Switzerland 2021 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s).