Analysis of Microbial Community Structure of Pit Mud for Chinese Strong-Flavor Liquor Fermentation Using Next Generation DNA Sequencing of Full-Length 16S Rrna
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

From Sporulation to Intracellular Offspring Production: Evolution
FROM SPORULATION TO INTRACELLULAR OFFSPRING PRODUCTION: EVOLUTION OF THE DEVELOPMENTAL PROGRAM OF EPULOPISCIUM A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by David Alan Miller January 2012 © 2012 David Alan Miller FROM SPORULATION TO INTRACELLULAR OFFSPRING PRODUCTION: EVOLUTION OF THE DEVELOPMENTAL PROGRAM OF EPULOPISCIUM David Alan Miller, Ph. D. Cornell University 2012 Epulopiscium sp. type B is an unusually large intestinal symbiont of the surgeonfish Naso tonganus. Unlike most other bacteria, Epulopiscium sp. type B has never been observed to undergo binary fission. Instead, to reproduce, it forms multiple intracellular offspring. We believe this process is related to endospore formation, an ancient and complex developmental process performed by certain members of the Firmicutes. Endospore formation has been studied for over 50 years and is best characterized in Bacillus subtilis. To study the evolution of endospore formation in the Firmicutes and the relatedness of this process to intracellular offspring formation in Epulopiscium, we have searched for sporulation genes from the B. subtilis model in all of the completed genomes of members of the Firmicutes, in addition to Epulopiscium sp. type B and its closest relative, the spore-forming Cellulosilyticum lentocellum. By determining the presence or absence of spore genes, we see the evolution of endospore formation in closely related bacteria within the Firmicutes and begin to predict if 19 previously characterized non-spore-formers have the genetic capacity to form a spore. We can also map out sporulation-specific mechanisms likely being used by Epulopiscium for offspring formation. -
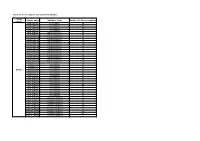
Table S1: List of Samples Included in the Analysis
Table S1: list of samples included in the analysis Study Sample name Inhibitory status Number of days at sampling number DNA.0P2T4 No inhibition 29 DNA.0P2T6 No inhibition 57 DNA.10P2T4 No inhibition 29 DNA.10P2T6 No inhibition 57 DNA.75P2T4 Phenol inhibition 29 DNA.75P2T6 Phenol inhibition 57 DNA.100P2T4 Phenol inhibition 29 DNA.100P2T6 Phenol inhibition 57 DNA.125P1T4 Phenol inhibition 29 DNA.125P1T6 Phenol inhibition 57 DNA.125P2T4 Phenol inhibition 29 DNA.125P2T6 Phenol inhibition 57 DNA.125P3T4 Phenol inhibition 29 DNA.125P3T6 Phenol inhibition 57 DNA.150P2T4 Phenol inhibition 29 DNA.150P2T6 Phenol inhibition 57 DNA.200P2T4 Phenol inhibition 29 DNA.200P2T6 Phenol inhibition 57 DNA.0N2T4 No inhibition 29 DNA.0N2T5 No inhibition 42 DNA.0N2T6 No inhibition 57 Study 1 DNA.5N2T4 No inhibition 29 DNA.5N2T5 No inhibition 42 DNA.5N2T6 No inhibition 57 DNA.10N2T4 No inhibition 29 DNA.10N2T5 No inhibition 42 DNA.10N2T6 No inhibition 57 DNA.15N2T4 No inhibition 29 DNA.15N2T5 No inhibition 42 DNA.15N2T6 No inhibition 57 DNA.25N2T4 No inhibition 29 DNA.25N2T5 No inhibition 42 DNA.25N2T6 No inhibition 57 DNA.75N2T4 Ammonia inhibition 29 DNA.75N2T5 Ammonia inhibition 42 DNA.75N2T6 Ammonia inhibition 57 DNA.100N2T4 Ammonia inhibition 29 DNA.100N2T5 Ammonia inhibition 42 DNA.100N2T6 Ammonia inhibition 57 DNA.250N2T4 Ammonia inhibition 29 DNA.250N2T5 Ammonia inhibition 42 DNA.250N2T6 Ammonia inhibition 57 nono2T3 No inhibition 16 noN2T4 Ammonia inhibition 23 noN2T8 Ammonia inhibition 60 noN2T9 Ammonia inhibition 85 noPhi2T4 Phenol inhibition 23 noPhi2T5 -

The Core Populations and Co-Occurrence Patterns Of
Rui et al. Biotechnol Biofuels (2015) 8:158 DOI 10.1186/s13068-015-0339-3 RESEARCH Open Access The core populations and co‑occurrence patterns of prokaryotic communities in household biogas digesters Junpeng Rui1,2†, Jiabao Li1,2†, Shiheng Zhang1,2, Xuefeng Yan1,2, Yuanpeng Wang3 and Xiangzhen Li1,2* Abstract Background: Household biogas digesters are widely used to harvest energy in rural areas of developing countries. Understanding core prokaryotic communities, their co-occurrence patterns, and their relationships to environmental factors is important to manage these small-scale anaerobic digestion systems effectively. In this study, 43 household biogas digesters were collected across eight provinces in China. Prokaryotic communities were investigated using 454 pyrosequencing of 16S rRNA genes. Results: Fourteen core genera and ten core OTUs were identified in household biogas digesters. They were mainly affiliated with the phylum Firmicutes, Synergistetes, Actinobacteria, Chloroflexi, and Spirochaetes. Core prokaryotic genera were mainly composed of Clostridium, Clostridium XI, Syntrophomonas, Cloacibacillus, Sedimentibacter, and Turicibacter. Prokaryotic communities in the 43 samples were clearly divided into two clusters. Cluster I was domi- nated by Clostridium, while Cluster II was dominated by members of Spirochaetes, Bacteroidales, Clostridia, and abundant syntrophs and methanogens. NH4+-N and COD contributed significantly to the assembly of the prokaryotic community in Cluster I, while NH4+-N, pH, and phosphate contributed significantly to Cluster II. Correlation-based network analysis showed that the prokaryotic communities in the biogas digesters were dominated by some func- tional modules. Cluster I was dominated by acetotrophic methanogenic modules and the Clostridium-driven primary fermentation module, while the network of Cluster II was dominated by hydrogenotrophic and acetogenic methano- genesis modules and multi-group-driven (Spirochaetes, Bacteroidales, and Clostridia) primary fermentation modules. -

Nutritional Features of Syntrophomonas Wolfei P
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Oct. 1990, p. 3223-3224 Vol. 56, No. 10 0099-2240/90/103223-02$02.00/0 Copyright © 1990, American Society for Microbiology Nutritional Features of Syntrophomonas wolfei P. SHAWN BEATY AND MICHAEL J. McINERNEY* Department ofBotany and Microbiology, University of Oklahoma, Norman, Oklahoma 73019-0245 Received 27 March 1990/Accepted 18 July 1990 Syntrophomonas wolfei subsp. wolfei grew poorly in a defined medium with crotonate as the energy source in the absence of rumen fluid. Thiamine, lipoic acid, biotin, cyanocobalamin, and para-aminobenzoic acid were required for growth comparable to that obtained with the rumen fluid-based medium. Iron and cobalt were also required for the growth of S. wolfei in the chemically defined medium. Propionate and longer-chain fatty acids are important in medium lacking a particular compound before it was intermediates in the conversion of organic matter to CH4 and concluded that the compound was not required for the CO2 (4). These compounds are degraded to methanogenic growth. Growth was monitored spectrophotometrically at substrates (acetate, H2, and formate) by syntrophic bacteria 600 nm, and growth rates were calculated from changes in which require the presence of methanogens or other H2- absorbance (6). using bacteria to maintain low concentrations of H2 for the The pure culture of S. wolfei grew at a specific growth rate degradation offatty acids to be thermodynamically favorable of 0.039/h in crotonate basal medium which contained rumen (4). Syntrophomonas wolfei subsp. wolfei is an anaerobic fluid. Decreasing the rumen fluid concentration of this me- bacterium that a-oxidizes short-chain saturated fatty acids, dium from 5 to 0.5% (vol/vol) did not affect the rate of four to eight carbons in length, to acetate, propionate (from growth or the final absorbance (0.45 to 0.5) of the culture odd-numbered fatty acids), and H2 only when grown in (data not shown). -

Research Article Archaea and Bacteria Acclimate to High Total Ammonia in a Methanogenic Reactor Treating Swine Waste
Hindawi Publishing Corporation Archaea Volume 2016, Article ID 4089684, 10 pages http://dx.doi.org/10.1155/2016/4089684 Research Article Archaea and Bacteria Acclimate to High Total Ammonia in a Methanogenic Reactor Treating Swine Waste Sofia Esquivel-Elizondo,1,2 Prathap Parameswaran,3 Anca G. Delgado,1 Juan Maldonado,1 Bruce E. Rittmann,1,2 and Rosa Krajmalnik-Brown1,2 1 Swette Center for Environmental Biotechnology, The Biodesign Institute, Arizona State University, P.O. Box 875701, Tempe, AZ 85287-5701, USA 2School of Sustainable Engineering and the Built Environment, Arizona State University, Tempe, AZ, USA 3Department of Civil Engineering, Kansas State University, 2118 Fiedler Hall, Manhattan, KS 66506, USA Correspondence should be addressed to Rosa Krajmalnik-Brown; [email protected] Received 10 June 2016; Accepted 11 August 2016 Academic Editor: Jessica A. Smith Copyright © 2016 Sofia Esquivel-Elizondo et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Inhibition by ammonium at concentrations above 1000 mgN/L is known to harm the methanogenesis phase of anaerobic digestion. We anaerobically digested swine waste and achieved steady state COD-removal efficiency of around 52% with no fatty-acid or H2 accumulation. As the anaerobic microbial community adapted to the gradual increase of total ammonia-N (NH3-N) from 890 ± 295 to 2040 ± 30 mg/L, the Bacterial and Archaeal communities became less diverse. Phylotypes most closely related to hydrogenotrophic Methanoculleus (36.4%) and Methanobrevibacter (11.6%), along with acetoclastic Methanosaeta (29.3%), became the most abundant Archaeal sequences during acclimation. -

Comparative Genomics of Syntrophic Branched-Chain Fatty Acid Degrading Bacteria
Microbes Environ. Vol. 31, No. 3, 288-292, 2016 https://www.jstage.jst.go.jp/browse/jsme2 doi:10.1264/jsme2.ME16057 Comparative Genomics of Syntrophic Branched-Chain Fatty Acid Degrading Bacteria TAKASHI NARIHIRO1,2†*, MASARU K. NOBU2†, HIDEYUKI TAMAKI1,3, YOICHI KAMAGATA1, YUJI SEKIGUCHI4, and WEN-TSO LIU2 1Bioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Central 6, Higashi 1–1–1, Tsukuba, Ibaraki 305–8566, Japan; 2Department of Civil and Environmental Engineering, University of Illinois at Urbana-Champaign, 205 North Mathews Ave, Urbana, IL 61801, USA; 3Biotechnology Research Institute, The University of Tokyo, 1–1–1 Yayoi, Bunkyo-ku, Tokyo 113–8657, Japan; and 4Biomedical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Central 6, Higashi 1–1–1, Tsukuba, Ibaraki 305–8566, Japan (Received March 16, 2016—Accepted June 5, 2016—Published online July 16, 2016) The syntrophic degradation of branched-chain fatty acids (BCFAs) such as 2-methylbutyrate and isobutyrate is an essential step in the production of methane from proteins/amino acids in anaerobic ecosystems. While a few syntrophic BCFA-degrading bacteria have been isolated, their metabolic pathways in BCFA and short-chain fatty acid (SCFA) degradation as well as energy conservation systems remain unclear. In an attempt to identify these pathways, we herein performed comparative genomics of three syntrophic bacteria: 2-methylbutyrate-degrading “Syntrophomonas wolfei subsp. methylbutyratica” strain JCM 14075T (=4J5T), isobutyrate-degrading Syntrophothermus lipocalidus strain TGB-C1T, and non-BCFA-metabolizing S. wolfei subsp. wolfei strain GöttingenT. We demonstrated that 4J5 and TGB-C1 both encode multiple genes/gene clusters involved in β-oxidation, as observed in the Göttingen genome, which has multiple copies of genes associated with butyrate degradation. -

Reversed Electron Transport in Syntrophic Degradation of Glucose, Butyrate and Ethanol
Reversed electron transport in syntrophic degradation of glucose, butyrate and ethanol Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) im Fachbereich Biologie der Mathematisch-Naturwissenschaftlichen Sektion der Universität Konstanz vorgelegt von Nicolai Müller Konstanz Februar 2010 Tag der mündlichen Prüfung : 09. April 2010 1. Referent : Prof. Dr. Bernhard Schink 2. Referent : Prof. Dr. Alasdair Cook 3. Referent : Prof. Dr. Wolfgang Buckel (Universität Marburg) Danksagung Die vorliegende Arbeit wurde im Zeitraum von Juni 2006 bis Januar 2010 am Lehrstuhl für Mikrobielle Ökologie von Prof. Dr. Bernhard Schink angefertigt. Mein besonderer Dank gilt Herrn Prof. Dr. Bernhard Schink für die Überlassung des Themas sowie sein stetes Interesse und seine ständige Diskussionsbereitschaft zu dieser Arbeit. Herrn Prof. Dr. Alasdair Cook danke ich für die Übernahme des Koreferates. PD Dr. Bodo Philipp danke ich für die zahlreichen methodischen Ratschläge und Diskussionen zu meiner Arbeit, insbesondere zu Beginn meiner Zeit als Doktorand. Mein Dank gilt ebenfalls Dr. David Schleheck, insbesondere für seine Kooperation und seinen Beitrag zum Manuskript über die syntrophe Oxidation von Butyrat nach der ersten Revision, aber auch für viele experimentelle Ratschläge und Diskussionsbereitschaft zum Butyratprojekt. Für ihre Hilfe, nicht nur bei der Optimierung der Proteinreinigung und SDS-PAGE, danke ich Karin Denger und Diliana Simeonova. Bei Antje Wiese bedanke ich mich insbesondere für ihre Unterstützung im Labor und die Herstellung der Kultivierungsmedien. Allen Mitgliedern der AG Schink und AG Cook, denen ich während meiner Zeit als Doktorand begegnet bin, danke ich für ihre Hilfsbereitschaft sowie eine sehr angenehme Arbeitsatmosphäre. Meinen Eltern danke ich herzlich für ihr Verständnis und ihre Unterstützung in all den Jahren meiner akademischen Ausbildung. -
DNA-SIP Based Genome-Centric Metagenomics Identifies Key Long-Chain Fatty Acid-Degrading Populations in Anaerobic Digesters with Different Feeding Frequencies
OPEN The ISME Journal (2018) 12, 112–123 www.nature.com/ismej ORIGINAL ARTICLE DNA-SIP based genome-centric metagenomics identifies key long-chain fatty acid-degrading populations in anaerobic digesters with different feeding frequencies Ryan M Ziels1,2, Diana Z Sousa3, H David Stensel2 and David AC Beck4,5 1Department of Civil Engineering, University of British Columbia, Vancouver, British Columbia, Canada; 2Department of Civil and Environmental Engineering, University of Washington, Seattle, WA, USA; 3Laboratory of Microbiology, Wageningen University, Wageningen, The Netherlands; 4eScience Institute, University of Washington, Seattle, WA, USA and 5Department of Chemical Engineering, University of Washington, Seattle, WA, USA Fats, oils and greases (FOG) are energy-dense wastes that can be added to anaerobic digesters to substantially increase biomethane recovery via their conversion through long-chain fatty acids (LCFAs). However, a better understanding of the ecophysiology of syntrophic LCFA-degrading microbial communities in anaerobic digesters is needed to develop operating strategies that mitigate inhibitory LCFA accumulation from FOG. In this research, DNA stable isotope probing (SIP) was coupled with metagenomic sequencing for a genome-centric comparison of oleate (C18:1)-degrading populations in two anaerobic codigesters operated with either a pulse feeding or continuous-feeding strategy. The pulse-fed codigester microcosms converted oleate into methane at over 20% higher rates than the continuous-fed codigester microcosms. Differential coverage binning was demon- strated for the first time to recover population genome bins (GBs) from DNA-SIP metagenomes. About 70% of the 13C-enriched GBs were taxonomically assigned to the Syntrophomonas genus, thus substantiating the importance of Syntrophomonas species to LCFA degradation in anaerobic digesters. -

Peat: Home to Novel Syntrophic Species That Feed Acetate- and Hydrogen-Scavenging Methanogens
The ISME Journal (2016) 10, 1954–1966 © 2016 International Society for Microbial Ecology All rights reserved 1751-7362/16 www.nature.com/ismej ORIGINAL ARTICLE Peat: home to novel syntrophic species that feed acetate- and hydrogen-scavenging methanogens Oliver Schmidt, Linda Hink, Marcus A Horn and Harold L Drake Department of Ecological Microbiology, University of Bayreuth, Bayreuth, Germany Syntrophic bacteria drive the anaerobic degradation of certain fermentation products (e.g., butyrate, ethanol, propionate) to intermediary substrates (e.g., H2, formate, acetate) that yield methane at the ecosystem level. However, little is known about the in situ activities and identities of these syntrophs in peatlands, ecosystems that produce significant quantities of methane. The consumption of butyrate, ethanol or propionate by anoxic peat slurries at 5 and 15 °C yielded methane and CO2 as the sole accumulating products, indicating that the intermediates H2, formate and acetate were scavenged effectively by syntrophic methanogenic consortia. 16S rRNA stable isotope probing identified novel species/strains of Pelobacter and Syntrophomonas that syntrophically oxidized ethanol and butyrate, respectively. Propionate was syntrophically oxidized by novel species of Syntrophobacter and Smithella, genera that use different propionate-oxidizing pathways. Taxa not known for a syntrophic metabolism may have been involved in the oxidation of butyrate (Telmatospirillum-related) and propionate (unclassified Bacteroidetes and unclassified Fibrobac- teres). Gibbs free energies (ΔGs) for syntrophic oxidations of ethanol and butyrate were more favorable than ΔGs for syntrophic oxidation of propionate. As a result of the thermodynamic constraints, acetate transiently accumulated in ethanol and butyrate treatments but not in propionate treatments. Aceticlastic methanogens (Methanosarcina, Methanosaeta) appeared to outnumber hydrogenotrophic methanogens (Methanocella, Methanoregula), reinforcing the likely importance of aceticlastic methanogenesis to the overall production of methane. -

Methane Production from Oleate: Assessing the Bioaugmentation Potential of Syntrophomonas Zehnderi
water research 44 (2010) 4940e4947 Available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/watres Methane production from oleate: Assessing the bioaugmentation potential of Syntrophomonas zehnderi A.J. Cavaleiro*, D.Z. Sousa, M.M. Alves Institute for Biotechnology and Bioengineering, Centre of Biological Engineering, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal article info abstract Article history: The potential for improving long-chain fatty acids (LCFA) conversion to methane was Received 2 April 2010 evaluated by bioaugmenting a non-acclimated anaerobic granular sludge with Syntropho- Received in revised form monas zehnderi. Batch bioaugmentation assays were performed with and without the solid 2 July 2010 microcarrier sepiolite, using 1 mM oleate as sole carbon and energy source. When S. Accepted 13 July 2010 zehnderi was added to the anaerobic sludge methane production from oleate was faster. Available online 21 July 2010 High methane yields, i.e. 89 Æ 5% and 72 Æ 1%, were observed in bioaugmented assays in the absence and presence of sepiolite, respectively. Sepiolite stimulated a faster methane Keywords: production from oleate and prevented the accumulation of acetate. Acetoclastic activity Bioaugmentation was affected by oleate in the absence of sepiolite, where methane production rate was 26% Syntrophomonas zehnderi lower than in assays with microcarrier. Methane ª 2010 Elsevier Ltd. All rights reserved. Oleate Sepiolite 1. Introduction possible (Stams, 1994). Presently, there are 7 species of syn- trophic bacteria reported as capable of growing on LCFA with Long-chain fatty acids (LCFA) are produced during the hydrolysis more than 12 carbon atoms. Among these bacteria only 3 of oils and fats and are commonly present in fatty-wastewaters. -

Microbial Ecology, Biogeochemistry, and Signatures of Life at Low Temperature in Arctic Thermokarst Lake Sediments and High Sierra Snow Fields
University of Nevada, Reno Microbial Ecology, Biogeochemistry, and Signatures of Life at Low Temperature in Arctic Thermokarst Lake Sediments and High Sierra Snow Fields A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biochemistry by Paula B. Matheus Carnevali Dr. Alison Murray/Dissertation Advisor May, 2015 Copyright by Paula B. Matheus Carnevali 2015 All Rights Reserved THE GRADUATE SCHOOL We recommend that the dissertation prepared under our supervision by PAULA B. MATHEUS CARNEVALI Entitled Microbial Ecology, Biogeochemistry, And Signatures Of Life At Low Temperature In Arctic Thermokarst Lake Sediments And High Sierra Snow Fields be accepted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Alison E. Murray, Ph.D., Advisor Gary Blomquist, Ph.D., Committee Member David Shintani, Ph.D., Committee Member Henry Sun, Ph.D., Committee Member Kevin Hand, Ph.D., Committee Member Jerry Qualls, Ph.D., Graduate School Representative David W. Zeh, Ph. D., Dean, Graduate School May, 2015 i Abstract The subjects of biological methane (CH4) production and microbial community diversity and structure in extreme cold environments were at the core of this dissertation. CH4 is a potent greenhouse gas that contributes to the warming of the planet and has chemical properties that allow its detection by instruments that can go into space. In this sense, CH4 is also a biosignature, because its presence could be indicative of the presence of life. Mechanisms that lead to the formation of this gas on Earth include reactions mediated by biological enzymes that evolved early in life’s evolutionary time.