From Sporulation to Intracellular Offspring Production: Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -
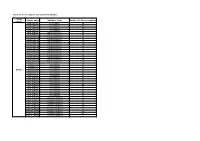
Table S1: List of Samples Included in the Analysis
Table S1: list of samples included in the analysis Study Sample name Inhibitory status Number of days at sampling number DNA.0P2T4 No inhibition 29 DNA.0P2T6 No inhibition 57 DNA.10P2T4 No inhibition 29 DNA.10P2T6 No inhibition 57 DNA.75P2T4 Phenol inhibition 29 DNA.75P2T6 Phenol inhibition 57 DNA.100P2T4 Phenol inhibition 29 DNA.100P2T6 Phenol inhibition 57 DNA.125P1T4 Phenol inhibition 29 DNA.125P1T6 Phenol inhibition 57 DNA.125P2T4 Phenol inhibition 29 DNA.125P2T6 Phenol inhibition 57 DNA.125P3T4 Phenol inhibition 29 DNA.125P3T6 Phenol inhibition 57 DNA.150P2T4 Phenol inhibition 29 DNA.150P2T6 Phenol inhibition 57 DNA.200P2T4 Phenol inhibition 29 DNA.200P2T6 Phenol inhibition 57 DNA.0N2T4 No inhibition 29 DNA.0N2T5 No inhibition 42 DNA.0N2T6 No inhibition 57 Study 1 DNA.5N2T4 No inhibition 29 DNA.5N2T5 No inhibition 42 DNA.5N2T6 No inhibition 57 DNA.10N2T4 No inhibition 29 DNA.10N2T5 No inhibition 42 DNA.10N2T6 No inhibition 57 DNA.15N2T4 No inhibition 29 DNA.15N2T5 No inhibition 42 DNA.15N2T6 No inhibition 57 DNA.25N2T4 No inhibition 29 DNA.25N2T5 No inhibition 42 DNA.25N2T6 No inhibition 57 DNA.75N2T4 Ammonia inhibition 29 DNA.75N2T5 Ammonia inhibition 42 DNA.75N2T6 Ammonia inhibition 57 DNA.100N2T4 Ammonia inhibition 29 DNA.100N2T5 Ammonia inhibition 42 DNA.100N2T6 Ammonia inhibition 57 DNA.250N2T4 Ammonia inhibition 29 DNA.250N2T5 Ammonia inhibition 42 DNA.250N2T6 Ammonia inhibition 57 nono2T3 No inhibition 16 noN2T4 Ammonia inhibition 23 noN2T8 Ammonia inhibition 60 noN2T9 Ammonia inhibition 85 noPhi2T4 Phenol inhibition 23 noPhi2T5 -

EXPERIMENTAL STUDIES on FERMENTATIVE FIRMICUTES from ANOXIC ENVIRONMENTS: ISOLATION, EVOLUTION, and THEIR GEOCHEMICAL IMPACTS By
EXPERIMENTAL STUDIES ON FERMENTATIVE FIRMICUTES FROM ANOXIC ENVIRONMENTS: ISOLATION, EVOLUTION, AND THEIR GEOCHEMICAL IMPACTS By JESSICA KEE EUN CHOI A dissertation submitted to the School of Graduate Studies Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Microbial Biology Written under the direction of Nathan Yee And approved by _______________________________________________________ _______________________________________________________ _______________________________________________________ _______________________________________________________ New Brunswick, New Jersey October 2017 ABSTRACT OF THE DISSERTATION Experimental studies on fermentative Firmicutes from anoxic environments: isolation, evolution and their geochemical impacts by JESSICA KEE EUN CHOI Dissertation director: Nathan Yee Fermentative microorganisms from the bacterial phylum Firmicutes are quite ubiquitous in subsurface environments and play an important biogeochemical role. For instance, fermenters have the ability to take complex molecules and break them into simpler compounds that serve as growth substrates for other organisms. The research presented here focuses on two groups of fermentative Firmicutes, one from the genus Clostridium and the other from the class Negativicutes. Clostridium species are well-known fermenters. Laboratory studies done so far have also displayed the capability to reduce Fe(III), yet the mechanism of this activity has not been investigated -

Development of a Metabolic Pathway Transfer and Genomic Integration
Philipps et al. Biotechnol Biofuels (2019) 12:112 https://doi.org/10.1186/s13068-019-1448-1 Biotechnology for Biofuels RESEARCH Open Access Development of a metabolic pathway transfer and genomic integration system for the syngas-fermenting bacterium Clostridium ljungdahlii Gabriele Philipps1, Sebastian de Vries1,2 and Stefan Jennewein1* Abstract Background: Clostridium spp. can synthesize valuable chemicals and fuels by utilizing diverse waste-stream sub- strates, including starchy biomass, lignocellulose, and industrial waste gases. However, metabolic engineering in Clostridium spp. is challenging due to the low efciency of gene transfer and genomic integration of entire biosyn- thetic pathways. Results: We have developed a reliable gene transfer and genomic integration system for the syngas-fermenting bacterium Clostridium ljungdahlii based on the conjugal transfer of donor plasmids containing large transgene cas- settes (> 5 kb) followed by the inducible activation of Himar1 transposase to promote integration. We established a conjugation protocol for the efcient generation of transconjugants using the Gram-positive origins of replica- tion repL and repH. We also investigated the impact of DNA methylation on conjugation efciency by testing donor constructs with all possible combinations of Dam and Dcm methylation patterns, and used bisulfte conversion and PacBio sequencing to determine the DNA methylation profle of the C. ljungdahlii genome, resulting in the detection of four sequence motifs with N6-methyladenosine. As proof of concept, we demonstrated the transfer and genomic integration of a heterologous acetone biosynthesis pathway using a Himar1 transposase system regulated by a xylose-inducible promoter. The functionality of the integrated pathway was confrmed by detecting enzyme proteo- typic peptides and the formation of acetone and isopropanol by C. -

Gene Conservation Among Endospore-Forming Bacteria Reveals Additional Sporulation Genes in Bacillus Subtilis
Gene Conservation among Endospore-Forming Bacteria Reveals Additional Sporulation Genes in Bacillus subtilis Bjorn A. Traag,a Antonia Pugliese,a Jonathan A. Eisen,b Richard Losicka Department of Molecular and Cellular Biology, Harvard University, Cambridge, Massachusetts, USAa; Department of Evolution and Ecology, University of California Davis Genome Center, Davis, California, USAb The capacity to form endospores is unique to certain members of the low-G؉C group of Gram-positive bacteria (Firmicutes) and requires signature sporulation genes that are highly conserved across members of distantly related genera, such as Clostridium and Bacillus. Using gene conservation among endospore-forming bacteria, we identified eight previously uncharacterized genes that are enriched among endospore-forming species. The expression of five of these genes was dependent on sporulation-specific transcription factors. Mutants of none of the genes exhibited a conspicuous defect in sporulation, but mutants of two, ylxY and ylyA, were outcompeted by a wild-type strain under sporulation-inducing conditions, but not during growth. In contrast, a ylmC mutant displayed a slight competitive advantage over the wild type specific to sporulation-inducing conditions. The phenotype of a ylyA mutant was ascribed to a defect in spore germination efficiency. This work demonstrates the power of combining phy- logenetic profiling with reverse genetics and gene-regulatory studies to identify unrecognized genes that contribute to a con- served developmental process. he formation of endospores is a distinctive developmental the successive actions of four compartment-specific sigma factors Tprocess wherein a dormant cell type (the endospore) is formed (appearing in the order F, E, G, and K), whose activities are inside another cell (the mother cell) and ultimately released into confined to the forespore (F and G) or the mother cell (E and the environment by lysis of the mother cell (1, 2). -

A Metagenomic Window Into the 2-Km-Deep Terrestrial Subsurface Aquifer Revealed Multiple Pathways of Organic Matter Decomposition Vitaly V
FEMS Microbiology Ecology, 94, 2018, fiy152 doi: 10.1093/femsec/fiy152 Advance Access Publication Date: 7 August 2018 Research Article RESEARCH ARTICLE A metagenomic window into the 2-km-deep terrestrial subsurface aquifer revealed multiple pathways of organic matter decomposition Vitaly V. Kadnikov1, Andrey V. Mardanov1, Alexey V. Beletsky1, David Banks3, Nikolay V. Pimenov4, Yulia A. Frank2,OlgaV.Karnachuk2 and Nikolai V. Ravin1,* 1Institute of Bioengineering, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky prosp. 33-2, Moscow, 119071, Russia, 2Laboratory of Biochemistry and Molecular Biology, Tomsk State University, Lenina prosp. 35, Tomsk, 634050, Russia, 3School of Engineering, Systems Power & Energy, Glasgow University, Glasgow G12 8QQ, and Holymoor Consultancy Ltd., 360 Ashgate Road, Chesterfield, Derbyshire S40 4BW, UK and 4Winogradsky Institute of Microbiology, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky prosp 33-2, Moscow, 119071, Russia ∗Corresponding author: Institute of Bioengineering, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky prosp. 33-2, Moscow, 119071, Russia. Tel: +74997833264; Fax: +74991353051; E-mail: [email protected] One sentence summary: metagenome-assembled genomes of microorganisms from the deep subsurface thermal aquifer revealed functional diversity of the community and novel uncultured bacterial lineages Editor: Tillmann Lueders ABSTRACT We have sequenced metagenome of the microbial community of a deep subsurface thermal aquifer in the Tomsk Region of the Western Siberia, Russia. Our goal was the recovery of near-complete genomes of the community members to enable accurate reconstruction of metabolism and ecological roles of the microbial majority, including previously unstudied lineages. The water, obtained via a 2.6 km deep borehole 1-R, was anoxic, with a slightly alkaline pH, and a temperature around 45◦C. -

ABSTRACT WHITHAM, JASON MICHAEL. Identifying Genetic
ABSTRACT WHITHAM, JASON MICHAEL. Identifying Genetic Targets and Growth Conditions for Higher Ethanol Production by the Synthesis Gas Fermenting Bacterium Clostridium ljungdahlii (Under direction of Dr. Amy M. Grunden and Dr. Joel J. Pawlak). The United States has increasing volume mandates for cellulosic biofuels production to reduce our dependence on foreign oil for the sake of transportation fuel security. For the past few years, mandated volumes have not been met by industry though for a variety of reasons. Clostridium ljungdahlii, an established cellulosic ethanol- producing industrial biocatalyst can produce ethanol from synthesis gas; however, relatively low concentrations of ethanol compared to traditional yeast fermentations of sugar are costly to separate from the fermentation medium, and C. ljungdahlii’s growth and productivity can be inhibited by contaminants in syngas which are expensive to remove. Synthesis gas is predominantly composed of carbon monoxide, carbon dioxide, and hydrogen, but also can contain a variety of contaminants (e.g. hydrogen cyanide, mono-nitrogen oxides, ammonia, sulfur dioxide, carbonyl sulfide, hydrogen sulfide, oxygen) depending on the carbonaceous feedstock from which it is produced (e.g. biomass, agricultural waste, municipal waste, industrial waste). The purpose of this work was, therefore, to identify genetic targets and growth conditions for higher ethanol production. To accomplish this, the metabolic and physiological response of C. ljungdahlii PETC to oxygen was investigated and a lab-derived hyper ethanol-producing strain of C. ljungdahlii (designated OTA1) was characterized. Preliminary investigation showed that C. ljungdahlii PETC has some tolerance to oxygen when cultured in a rich mixotrophic medium. Batch cultures not only continued to grow and consume H2, CO, and fructose after 8% O2 exposure, but fermentation product analysis revealed an increase in ethanol yield and decreased acetate yield compared to non-oxygen exposed cultures. -

Genome Diversity of Spore-Forming Firmicutes MICHAEL Y
Genome Diversity of Spore-Forming Firmicutes MICHAEL Y. GALPERIN National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894 ABSTRACT Formation of heat-resistant endospores is a specific Vibrio subtilis (and also Vibrio bacillus), Ferdinand Cohn property of the members of the phylum Firmicutes (low-G+C assigned it to the genus Bacillus and family Bacillaceae, Gram-positive bacteria). It is found in representatives of four specifically noting the existence of heat-sensitive vegeta- different classes of Firmicutes, Bacilli, Clostridia, Erysipelotrichia, tive cells and heat-resistant endospores (see reference 1). and Negativicutes, which all encode similar sets of core sporulation fi proteins. Each of these classes also includes non-spore-forming Soon after that, Robert Koch identi ed Bacillus anthracis organisms that sometimes belong to the same genus or even as the causative agent of anthrax in cattle and the species as their spore-forming relatives. This chapter reviews the endospores as a means of the propagation of this orga- diversity of the members of phylum Firmicutes, its current taxon- nism among its hosts. In subsequent studies, the ability to omy, and the status of genome-sequencing projects for various form endospores, the specific purple staining by crystal subgroups within the phylum. It also discusses the evolution of the violet-iodine (Gram-positive staining, reflecting the pres- Firmicutes from their apparently spore-forming common ancestor ence of a thick peptidoglycan layer and the absence of and the independent loss of sporulation genes in several different lineages (staphylococci, streptococci, listeria, lactobacilli, an outer membrane), and the relatively low (typically ruminococci) in the course of their adaptation to the saprophytic less than 50%) molar fraction of guanine and cytosine lifestyle in a nutrient-rich environment. -

Analysis of Microbial Community Structure of Pit Mud for Chinese Strong-Flavor Liquor Fermentation Using Next Generation DNA Sequencing of Full-Length 16S Rrna
bioRxiv preprint doi: https://doi.org/10.1101/380949; this version posted July 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Analysis of microbial community structure of pit mud for Chinese strong-flavor liquor fermentation using next generation DNA sequencing of full-length 16S rRNA Zuolin Liu1†, Ying Han1†, Junwei Li1,2, Runjie Cao2, Hongkui He2*, Anjun Li2 and Zhizhou Zhang1* 1School of Chemical Engineering and Chemistry, Harbin Institute of Technology, Harbin, China 150006 2The Center for Solid-state Fermentation Engineering of Anhui Province, The Anhui GujingTribute Liquor Ltd, Bozhou, Anhui, China, 236800 † *The corresponding author: [email protected]; [email protected] Equal contribution Contact: Zhizhou Zhang, PhD Prof [email protected] 86-631-5683176 Abstract. The pit is the necessary bioreactor for brewing process of Chinese strong-flavor liquor. Pit mud in pits contains a large number of microorganisms and is a complex ecosystem. The analysis of bacterial flora in pit mud is of great significance to understand liquor fermentation mechanisms. To overcome taxonomic limitations of short reads in 16S rRNA variable region sequencing, we used high-throughput DNA sequencing of near full-length 16S rRNA gene to analyze microbial compositions of different types of pit mud that produce different qualities of strong-flavor liquor. The results showed that the main species in pit mud were Pseudomonas extremaustralis 14-3, Pseudomonas veronii, Serratia marcescens WW4, and Clostridium leptum in Ruminiclostridium. -

The Core Populations and Co-Occurrence Patterns Of
Rui et al. Biotechnol Biofuels (2015) 8:158 DOI 10.1186/s13068-015-0339-3 RESEARCH Open Access The core populations and co‑occurrence patterns of prokaryotic communities in household biogas digesters Junpeng Rui1,2†, Jiabao Li1,2†, Shiheng Zhang1,2, Xuefeng Yan1,2, Yuanpeng Wang3 and Xiangzhen Li1,2* Abstract Background: Household biogas digesters are widely used to harvest energy in rural areas of developing countries. Understanding core prokaryotic communities, their co-occurrence patterns, and their relationships to environmental factors is important to manage these small-scale anaerobic digestion systems effectively. In this study, 43 household biogas digesters were collected across eight provinces in China. Prokaryotic communities were investigated using 454 pyrosequencing of 16S rRNA genes. Results: Fourteen core genera and ten core OTUs were identified in household biogas digesters. They were mainly affiliated with the phylum Firmicutes, Synergistetes, Actinobacteria, Chloroflexi, and Spirochaetes. Core prokaryotic genera were mainly composed of Clostridium, Clostridium XI, Syntrophomonas, Cloacibacillus, Sedimentibacter, and Turicibacter. Prokaryotic communities in the 43 samples were clearly divided into two clusters. Cluster I was domi- nated by Clostridium, while Cluster II was dominated by members of Spirochaetes, Bacteroidales, Clostridia, and abundant syntrophs and methanogens. NH4+-N and COD contributed significantly to the assembly of the prokaryotic community in Cluster I, while NH4+-N, pH, and phosphate contributed significantly to Cluster II. Correlation-based network analysis showed that the prokaryotic communities in the biogas digesters were dominated by some func- tional modules. Cluster I was dominated by acetotrophic methanogenic modules and the Clostridium-driven primary fermentation module, while the network of Cluster II was dominated by hydrogenotrophic and acetogenic methano- genesis modules and multi-group-driven (Spirochaetes, Bacteroidales, and Clostridia) primary fermentation modules. -

One-Step Production of C6–C8 Carboxylates by Mixed Culture Solely Grown on CO
He et al. Biotechnol Biofuels (2018) 11:4 https://doi.org/10.1186/s13068-017-1005-8 Biotechnology for Biofuels RESEARCH Open Access One‑step production of C6–C8 carboxylates by mixed culture solely grown on CO Pinjing He1,2,3, Wenhao Han1, Liming Shao2,3 and Fan Lü1,2,4* Abstract Background: This study aimed at producing C6–C8 medium-chain carboxylates (MCCAs) directly from gaseous CO using mixed culture. The yield and C2–C8 product composition were investigated when CO was continuously fed with gradually increasing partial pressure. Results: The maximal concentrations of n-caproate, n-heptylate, and n-caprylate were 1.892, 1.635, and 1 1 1 1.033 mmol L− , which were achieved at the maximal production rates of 0.276, 0.442, and 0.112 mmol L− day− , respectively. Microbial analysis revealed that long-term acclimation and high CO partial pressure were important to establish a CO-tolerant and CO-utilizing chain-elongating microbiome, rich in Acinetobacter, Alcaligenes, and Rhodo- bacteraceae and capable of forming MCCAs solely from CO. Conclusions: These results demonstrated that carboxylate and syngas platform could be integrated in a shared growth vessel, and could be a promising one-step technique to convert gaseous syngas to preferable liquid biochem- icals, thereby avoiding the necessity to coordinate syngas fermentation to short-chain carboxylates and short-to- medium-chain elongation. Thus, this method could provide an alternative solution for the utilization of waste-derived syngas and expand the resource of promising biofuels. Keywords: Syngas fermentation, Chain elongation, Carboxylates, One-step production, Mixed culture Background the thermal treatment of biowaste or artifcial polymers In recent times, conversion of waste to biochemicals [3–5]. -

Insights Into Key Parameters for Bio-Alcohol Production in Syngas Fermentation Using Model Carboxydotrophic Bacteria
INSIGHTS INTO KEY PARAMETERS FOR BIO-ALCOHOL PRODUCTION IN SYNGAS FERMENTATION USING MODEL CARBOXYDOTROPHIC BACTERIA Sara Ramió Pujol Per citar o enllaçar aquest document: Para citar o enlazar este documento: Use this url to cite or link to this publication: http://hdl.handle.net/10803/388041 ADVERTIMENT. L'accés als continguts d'aquesta tesi doctoral i la seva utilització ha de respectar els drets de la persona autora. Pot ser utilitzada per a consulta o estudi personal, així com en activitats o materials d'investigació i docència en els termes establerts a l'art. 32 del Text Refós de la Llei de Propietat Intel·lectual (RDL 1/1996). Per altres utilitzacions es requereix l'autorització prèvia i expressa de la persona autora. En qualsevol cas, en la utilització dels seus continguts caldrà indicar de forma clara el nom i cognoms de la persona autora i el títol de la tesi doctoral. No s'autoritza la seva reproducció o altres formes d'explotació efectuades amb finalitats de lucre ni la seva comunicació pública des d'un lloc aliè al servei TDX. Tampoc s'autoritza la presentació del seu contingut en una finestra o marc aliè a TDX (framing). Aquesta reserva de drets afecta tant als continguts de la tesi com als seus resums i índexs. ADVERTENCIA. El acceso a los contenidos de esta tesis doctoral y su utilización debe respetar los derechos de la persona autora. Puede ser utilizada para consulta o estudio personal, así como en actividades o materiales de investigación y docencia en los términos establecidos en el art.