The Core Populations and Co-Occurrence Patterns Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

From Sporulation to Intracellular Offspring Production: Evolution
FROM SPORULATION TO INTRACELLULAR OFFSPRING PRODUCTION: EVOLUTION OF THE DEVELOPMENTAL PROGRAM OF EPULOPISCIUM A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by David Alan Miller January 2012 © 2012 David Alan Miller FROM SPORULATION TO INTRACELLULAR OFFSPRING PRODUCTION: EVOLUTION OF THE DEVELOPMENTAL PROGRAM OF EPULOPISCIUM David Alan Miller, Ph. D. Cornell University 2012 Epulopiscium sp. type B is an unusually large intestinal symbiont of the surgeonfish Naso tonganus. Unlike most other bacteria, Epulopiscium sp. type B has never been observed to undergo binary fission. Instead, to reproduce, it forms multiple intracellular offspring. We believe this process is related to endospore formation, an ancient and complex developmental process performed by certain members of the Firmicutes. Endospore formation has been studied for over 50 years and is best characterized in Bacillus subtilis. To study the evolution of endospore formation in the Firmicutes and the relatedness of this process to intracellular offspring formation in Epulopiscium, we have searched for sporulation genes from the B. subtilis model in all of the completed genomes of members of the Firmicutes, in addition to Epulopiscium sp. type B and its closest relative, the spore-forming Cellulosilyticum lentocellum. By determining the presence or absence of spore genes, we see the evolution of endospore formation in closely related bacteria within the Firmicutes and begin to predict if 19 previously characterized non-spore-formers have the genetic capacity to form a spore. We can also map out sporulation-specific mechanisms likely being used by Epulopiscium for offspring formation. -
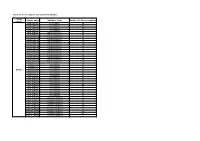
Table S1: List of Samples Included in the Analysis
Table S1: list of samples included in the analysis Study Sample name Inhibitory status Number of days at sampling number DNA.0P2T4 No inhibition 29 DNA.0P2T6 No inhibition 57 DNA.10P2T4 No inhibition 29 DNA.10P2T6 No inhibition 57 DNA.75P2T4 Phenol inhibition 29 DNA.75P2T6 Phenol inhibition 57 DNA.100P2T4 Phenol inhibition 29 DNA.100P2T6 Phenol inhibition 57 DNA.125P1T4 Phenol inhibition 29 DNA.125P1T6 Phenol inhibition 57 DNA.125P2T4 Phenol inhibition 29 DNA.125P2T6 Phenol inhibition 57 DNA.125P3T4 Phenol inhibition 29 DNA.125P3T6 Phenol inhibition 57 DNA.150P2T4 Phenol inhibition 29 DNA.150P2T6 Phenol inhibition 57 DNA.200P2T4 Phenol inhibition 29 DNA.200P2T6 Phenol inhibition 57 DNA.0N2T4 No inhibition 29 DNA.0N2T5 No inhibition 42 DNA.0N2T6 No inhibition 57 Study 1 DNA.5N2T4 No inhibition 29 DNA.5N2T5 No inhibition 42 DNA.5N2T6 No inhibition 57 DNA.10N2T4 No inhibition 29 DNA.10N2T5 No inhibition 42 DNA.10N2T6 No inhibition 57 DNA.15N2T4 No inhibition 29 DNA.15N2T5 No inhibition 42 DNA.15N2T6 No inhibition 57 DNA.25N2T4 No inhibition 29 DNA.25N2T5 No inhibition 42 DNA.25N2T6 No inhibition 57 DNA.75N2T4 Ammonia inhibition 29 DNA.75N2T5 Ammonia inhibition 42 DNA.75N2T6 Ammonia inhibition 57 DNA.100N2T4 Ammonia inhibition 29 DNA.100N2T5 Ammonia inhibition 42 DNA.100N2T6 Ammonia inhibition 57 DNA.250N2T4 Ammonia inhibition 29 DNA.250N2T5 Ammonia inhibition 42 DNA.250N2T6 Ammonia inhibition 57 nono2T3 No inhibition 16 noN2T4 Ammonia inhibition 23 noN2T8 Ammonia inhibition 60 noN2T9 Ammonia inhibition 85 noPhi2T4 Phenol inhibition 23 noPhi2T5 -

Pipeline for Amplifying and Analyzing Amplicons of the V1–V3 Region of the 16S Rrna Gene Heather K
Allen et al. BMC Res Notes (2016) 9:380 DOI 10.1186/s13104-016-2172-6 BMC Research Notes TECHNICAL NOTE Open Access Pipeline for amplifying and analyzing amplicons of the V1–V3 region of the 16S rRNA gene Heather K. Allen1*, Darrell O. Bayles2, Torey Looft1, Julian Trachsel1,3, Benjamin E. Bass1,4, David P. Alt2, Shawn M. D. Bearson1, Tracy Nicholson5 and Thomas A. Casey1 Abstract Background: Profiling of 16S rRNA gene sequences is an important tool for testing hypotheses in complex microbial communities, and analysis methods must be updated and validated as sequencing technologies advance. In host- associated bacterial communities, the V1–V3 region of the 16S rRNA gene is a valuable region to profile because it provides a useful level of taxonomic resolution; however, use of Illumina MiSeq data for experiments targeting this region needs validation. Results: Using a MiSeq machine and the version 3 (300 2) chemistry, we sequenced the V1–V3 region of the 16S rRNA gene within a mock community. Nineteen bacteria× and one archaeon comprised the mock community, and 12 replicate amplifications of the community were performed and sequenced. Sequencing the large fragment (490 bp) that encompasses V1–V3 yielded a higher error rate (3.6 %) than has been reported when using smaller fragment sizes. This higher error rate was due to a large number of sequences that occurred only one or two times among all mock community samples. Removing sequences that occurred one time among all samples (singletons) reduced the error rate to 1.4 %. Diversity estimates of the mock community containing all sequences were inflated, whereas estimates following singleton removal more closely reflected the actual mock community membership. -

ROLE of NOVEL QUORUM SENSING MOLECULES (DKPS-DIKETOPIPERAZINES) AS ACTIVATORS of BACTERIAL VIRULENCE and HOST RESPONSE Alex Gill
ROLE OF NOVEL QUORUM SENSING MOLECULES (DKPS-DIKETOPIPERAZINES) AS ACTIVATORS OF BACTERIAL VIRULENCE AND HOST RESPONSE Alex Gillone A thesis submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Master of Science in the Department of Periodontology in the School of Dentistry. Chapel Hill 2016 Approved by: Steven Offenbacher Silvana P. Barros Roland Arnold © 2016 Alex Gillone ALL RIGHTS RESERVED ii ABSTRACT Alex Gillone: Role of Novel Quorum sensing molecules (DKPs-Diketopiperazines) as activators of bacterial virulence and host response. (Under the direction of Steven Offenbacher) Objectives: The aim of this project was to establish the functional role of novel quorum sensing molecules (Diketopiperazines – DKPs) on activation of bacterial virulence properties and the potential effects on host cells as activators of the innate immune response. Methods: The effect of DKPs on the growth and virulence properties of the periodontal pathogen, Porphyromonas gingivalis (P.g.) A7436 strain was examined. Secondarily, the effect of DKPs on human monocyte (THP-1) viability, growth and cytokine production upon lipopolysaccharide (LPS) stimulation was determined. THP-1 cells were collected, counted and the cell lysate was evaluated for Interleukin 1β (IL-1β) mRNA expression. Results: Our results demonstrate that DKPs minimally affect the growth of P. g. DKP alone did not significantly alter THP-1 viability (p=0.20), indicating it was not toxic to the cells. However, analysis of the IL-1β mRNA expression indicates that DKP inhibited the inflammatory response of LPS-stimulated THP-1 cells. Conclusions: We have confirmed that DKPs minimally affect the growth of P. -

This Is an Open Access-Journal's PDF Published in Chertkov, O., Sikorski
Complete genome sequence of Aminobacterium colombiense type strain (ALA-1T) Authors Chertkov, Olga; Sikorski, Johannes; Brambilla, Evelyne; Lapidus, Alla; Copeland, Alex; Glavina Del Rio, Tijana; Nolan, Matt; Lucas, Susan; Tice, Hope; Cheng, Jan-Fang; Han, Cliff; Detter, John C.; Bruce, David; Tapia, Roxanne; Goodwin, Lynne; Pitluck, Sam; Liolios, Konstantinos; Ivanova, Natalia; Mavromatis, Konstantinos; Ovchinnikova, Galina; Pati, Amrita; Chen, Amy; Palaniappan, Krishna; Land, Miriam; Hauser, Loren; Chang, Yun- Juan; Jeffries, Cynthia D.; Spring, Stefan; Rohde, Manfred; Göker, Markus; Bristow, James; Eisen, Jonathan A.; Markowitz, Victor; Hugenholtz, Philip; Kyrpides, Nikos C.; Klenk, Hans-Peter Citation Complete genome sequence of Aminobacterium colombiense type strain (ALA-1T) 2010, 2 (3):280 Standards in Genomic Sciences DOI 10.4056/sigs.902116 Journal Standards in Genomic Sciences Rights Archived with thanks to Standards in Genomic Sciences Download date 26/09/2021 00:40:08 Link to Item http://hdl.handle.net/2384/297243 This is an Open Access-journal’s PDF published in Chertkov, O., Sikorski, J., Brambilla, E., Lapidus, A., Copeland, A., del Rio, T.G., Nolan, M., Lucas, S., Tice, H., Cheng, J.-F., Han, C., Detter, J.C., Bruce, D., Tapia, R., Goodwin, L., Pitluck, S., Liolios, K., Ivanova, N., Mavromatis, K., Ovchinnikova, G., Pati, A., Chen, A., Palaniappan, K., Land, M., Hauser, L., Chang, Y.-J., Jeffries, C.D., Spring, S., Rohde, M., Göker, M., Bristow, J., Eisen, J.A., Markowitz, V., Hugenholtz, P., Kyrpides, N.C., Klenk, H.-P. Complete genome sequence of Aminobacterium colombiense type strain (ALA-1T) (2010) Standards in Genomic Sciences, 2 (3), pp. 280-289 Standards in Genomic Sciences (2010) 2:280-289 DOI:10.4056/sigs.902116 Complete genome sequence of Aminobacterium T colombiense type strain (ALA-1 ) Olga Chertkov1,2, Johannes Sikorski3, Evelyne Brambilla3, Alla Lapidus1, Alex Copeland1, Tijana Glavina Del Rio1, Matt Nolan1, Susan Lucas1, Hope Tice1, Jan-Fang Cheng1, Cliff Han1,4, John C. -

Analysis of Microbial Community Structure of Pit Mud for Chinese Strong-Flavor Liquor Fermentation Using Next Generation DNA Sequencing of Full-Length 16S Rrna
bioRxiv preprint doi: https://doi.org/10.1101/380949; this version posted July 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Analysis of microbial community structure of pit mud for Chinese strong-flavor liquor fermentation using next generation DNA sequencing of full-length 16S rRNA Zuolin Liu1†, Ying Han1†, Junwei Li1,2, Runjie Cao2, Hongkui He2*, Anjun Li2 and Zhizhou Zhang1* 1School of Chemical Engineering and Chemistry, Harbin Institute of Technology, Harbin, China 150006 2The Center for Solid-state Fermentation Engineering of Anhui Province, The Anhui GujingTribute Liquor Ltd, Bozhou, Anhui, China, 236800 † *The corresponding author: [email protected]; [email protected] Equal contribution Contact: Zhizhou Zhang, PhD Prof [email protected] 86-631-5683176 Abstract. The pit is the necessary bioreactor for brewing process of Chinese strong-flavor liquor. Pit mud in pits contains a large number of microorganisms and is a complex ecosystem. The analysis of bacterial flora in pit mud is of great significance to understand liquor fermentation mechanisms. To overcome taxonomic limitations of short reads in 16S rRNA variable region sequencing, we used high-throughput DNA sequencing of near full-length 16S rRNA gene to analyze microbial compositions of different types of pit mud that produce different qualities of strong-flavor liquor. The results showed that the main species in pit mud were Pseudomonas extremaustralis 14-3, Pseudomonas veronii, Serratia marcescens WW4, and Clostridium leptum in Ruminiclostridium. -

Draft Genome Sequence of Dethiosulfovibrio Salsuginis DSM
Díaz-Cárdenas et al. Standards in Genomic Sciences (2017) 12:86 DOI 10.1186/s40793-017-0303-x EXTENDEDGENOMEREPORT Open Access Draft genome sequence of Dethiosulfovibrio salsuginis DSM 21565T an anaerobic, slightly halophilic bacterium isolated from a Colombian saline spring Carolina Díaz-Cárdenas1, Gina López1, José David Alzate-Ocampo2, Laura N. González2, Nicole Shapiro3, Tanja Woyke3, Nikos C. Kyrpides3, Silvia Restrepo2 and Sandra Baena1* Abstract A bacterium belonging to the phylum Synergistetes, genus Dethiosulfovibrio was isolated in 2007 from a saline spring in Colombia. Dethiosulfovibrio salsuginis USBA 82T (DSM 21565T= KCTC 5659T) is a mesophilic, strictly anaerobic, slightly halophilic, Gram negative bacterium with a diderm cell envelope. The strain ferments peptides, amino acids and a few organic acids. Here we present the description of the complete genome sequencing and annotation of the type species Dethiosulfovibrio salsuginis USBA 82T. The genome consisted of 2.68 Mbp with a 53. 7% G + C. A total of 2609 genes were predicted and of those, 2543 were protein coding genes and 66 were RNA genes. We detected in USBA 82T genome six Synergistetes conserved signature indels (CSIs), specific for Jonquetella, Pyramidobacter and Dethiosulfovibrio. The genome of D. salsuginis contained, as expected, genes related to amino acid transport, amino acid metabolism and thiosulfate reduction. These genes represent the major gene groups of Synergistetes, related with their phenotypic traits, and interestingly, 11.8% of the genes in the genome belonged to the amino acid fermentation COG category. In addition, we identified in the genome some ammonification genes such as nitrate reductase genes. The presence of proline operon genes could be related to de novo synthesis of proline to protect the cell in response to high osmolarity. -

Nutritional Features of Syntrophomonas Wolfei P
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Oct. 1990, p. 3223-3224 Vol. 56, No. 10 0099-2240/90/103223-02$02.00/0 Copyright © 1990, American Society for Microbiology Nutritional Features of Syntrophomonas wolfei P. SHAWN BEATY AND MICHAEL J. McINERNEY* Department ofBotany and Microbiology, University of Oklahoma, Norman, Oklahoma 73019-0245 Received 27 March 1990/Accepted 18 July 1990 Syntrophomonas wolfei subsp. wolfei grew poorly in a defined medium with crotonate as the energy source in the absence of rumen fluid. Thiamine, lipoic acid, biotin, cyanocobalamin, and para-aminobenzoic acid were required for growth comparable to that obtained with the rumen fluid-based medium. Iron and cobalt were also required for the growth of S. wolfei in the chemically defined medium. Propionate and longer-chain fatty acids are important in medium lacking a particular compound before it was intermediates in the conversion of organic matter to CH4 and concluded that the compound was not required for the CO2 (4). These compounds are degraded to methanogenic growth. Growth was monitored spectrophotometrically at substrates (acetate, H2, and formate) by syntrophic bacteria 600 nm, and growth rates were calculated from changes in which require the presence of methanogens or other H2- absorbance (6). using bacteria to maintain low concentrations of H2 for the The pure culture of S. wolfei grew at a specific growth rate degradation offatty acids to be thermodynamically favorable of 0.039/h in crotonate basal medium which contained rumen (4). Syntrophomonas wolfei subsp. wolfei is an anaerobic fluid. Decreasing the rumen fluid concentration of this me- bacterium that a-oxidizes short-chain saturated fatty acids, dium from 5 to 0.5% (vol/vol) did not affect the rate of four to eight carbons in length, to acetate, propionate (from growth or the final absorbance (0.45 to 0.5) of the culture odd-numbered fatty acids), and H2 only when grown in (data not shown). -

Host-Associated Bacterial Taxa from Chlorobi, Chloroflexi, GN02, Synergistetes, SR1, TM7, and WPS-2 Phyla/Candidate Divisions
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Directory of Open Access Journals ournal of ralr æ icrobiologyi ORIGINAL ARTICLE Host-associated bacterial taxa from Chlorobi, Chloroflexi, GN02, Synergistetes, SR1, TM7, and WPS-2 Phyla/candidate divisions Anuj Camanocha1 and Floyd E. Dewhirst1,2* 1Department of Oral Medicine, Infection and Immunity, Harvard School of Dental Medicine, Boston, MA, USA; 2Department of Microbiology, The Forsyth Institute, Cambridge, MA, USA Background and objective: In addition to the well-known phyla Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, Spirochaetes, Fusobacteria, Tenericutes, and Chylamydiae, the oral microbiomes of mammals contain species from the lesser-known phyla or candidate divisions, including Synergistetes, TM7, Chlorobi, Chloroflexi, GN02, SR1, and WPS-2. The objectives of this study were to create phyla-selective 16S rDNA PCR primer pairs, create selective 16S rDNA clone libraries, identify novel oral taxa, and update canine and human oral microbiome databases. Design: 16S rRNA gene sequences for members of the lesser-known phyla were downloaded from GenBank and Greengenes databases and aligned with sequences in our RNA databases. Primers with potential phylum level selectivity were designed heuristically with the goal of producing nearly full-length 16S rDNA amplicons. The specificity of primer pairs was examined by making clone libraries from PCR amplicons and determining phyla identity by BLASTN analysis. Results: Phylum-selective primer pairs were identified that allowed construction of clone libraries with 96Á100% specificity for each of the lesser-known phyla. From these clone libraries, seven human and two canine novel oral taxa were identified and added to their respective taxonomic databases. -

Research Article Archaea and Bacteria Acclimate to High Total Ammonia in a Methanogenic Reactor Treating Swine Waste
Hindawi Publishing Corporation Archaea Volume 2016, Article ID 4089684, 10 pages http://dx.doi.org/10.1155/2016/4089684 Research Article Archaea and Bacteria Acclimate to High Total Ammonia in a Methanogenic Reactor Treating Swine Waste Sofia Esquivel-Elizondo,1,2 Prathap Parameswaran,3 Anca G. Delgado,1 Juan Maldonado,1 Bruce E. Rittmann,1,2 and Rosa Krajmalnik-Brown1,2 1 Swette Center for Environmental Biotechnology, The Biodesign Institute, Arizona State University, P.O. Box 875701, Tempe, AZ 85287-5701, USA 2School of Sustainable Engineering and the Built Environment, Arizona State University, Tempe, AZ, USA 3Department of Civil Engineering, Kansas State University, 2118 Fiedler Hall, Manhattan, KS 66506, USA Correspondence should be addressed to Rosa Krajmalnik-Brown; [email protected] Received 10 June 2016; Accepted 11 August 2016 Academic Editor: Jessica A. Smith Copyright © 2016 Sofia Esquivel-Elizondo et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Inhibition by ammonium at concentrations above 1000 mgN/L is known to harm the methanogenesis phase of anaerobic digestion. We anaerobically digested swine waste and achieved steady state COD-removal efficiency of around 52% with no fatty-acid or H2 accumulation. As the anaerobic microbial community adapted to the gradual increase of total ammonia-N (NH3-N) from 890 ± 295 to 2040 ± 30 mg/L, the Bacterial and Archaeal communities became less diverse. Phylotypes most closely related to hydrogenotrophic Methanoculleus (36.4%) and Methanobrevibacter (11.6%), along with acetoclastic Methanosaeta (29.3%), became the most abundant Archaeal sequences during acclimation. -

Metataxonomic and Histopathological Study of Rabbit Epizootic Enteropathy in Mexico
animals Article Metataxonomic and Histopathological Study of Rabbit Epizootic Enteropathy in Mexico Xiao-Haitzi Daniel Puón-Peláez 1, Neil Ross McEwan 2 , José Guadalupe Gómez-Soto 3 , Roberto Carlos Álvarez-Martínez 4 and Andrea Margarita Olvera-Ramírez 5,* 1 Doctorado en Ciencias Biológicas, Facultad de Ciencias Naturales, Universidad Autónoma de Querétaro, Avenida de las Ciencias S/N Juriquilla, Delegación Santa Rosa Jáuregui, Santiago de Querétaro, Qro. C.P. 76230, Mexico; [email protected] 2 School of Pharmacy & Life Sciences, Robert Gordon University, Garthdee Road, Aberdeen AB10 7GJ, UK; [email protected] 3 Cuerpo Académico de Nutrición y Reproducción Animal, Facultad de Ciencias Naturales, Universidad Autónoma de Querétaro, Avenida de las Ciencias S/N Juriquilla, Delegación Santa Rosa Jáuregui, Santiago de Querétaro, Qro. C.P. 76230, Mexico; [email protected] 4 Licenciatura en Microbiología, Facultad de Ciencias Naturales, Universidad Autónoma de Querétaro, Av. Junipero Serra, Antiguo Aeropuerto, Campus Aeropuerto S/N. Santiago de Querétaro, Qro. C.P. 76140, Mexico; [email protected] 5 Cuerpo Académico Salud Animal y Microbiología Ambiental, Facultad de Ciencias Naturales, Universidad Autónoma de Querétaro, Avenida de las Ciencias S/N Juriquilla, Delegación Santa Rosa Jáuregui, Santiago de Querétaro C.P. 76230, Mexico * Correspondence: [email protected]; Tel.: +52-442-192-1200 (ext. 5316) Received: 21 April 2020; Accepted: 24 May 2020; Published: 28 May 2020 Simple Summary: Epizootic rabbit enteropathy (ERE) is a worldwide-distributed dysbiotic syndrome that affects young rabbits. In Mexico, ERE represents 32% of the enteropathies that occur in rabbit production farms. The etiology of this syndrome has not been clarified yet; however, it has been associated with nutritional, environmental, and microbial factors.