Mercury Methylation by Metabolically Versatile and Cosmopolitan Marine Bacteria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exploring Antibiotic Susceptibility, Resistome and Mobilome Structure of Planctomycetes from Gemmataceae Family
sustainability Article Exploring Antibiotic Susceptibility, Resistome and Mobilome Structure of Planctomycetes from Gemmataceae Family Anastasia A. Ivanova *, Kirill K. Miroshnikov and Igor Y. Oshkin Research Center of Biotechnology of the Russian Academy of Sciences, Winogradsky Institute of Microbiology, 119071 Moscow, Russia; [email protected] (K.K.M.); [email protected] (I.Y.O.) * Correspondence: [email protected] Abstract: The family Gemmataceae accomodates aerobic, chemoorganotrophic planctomycetes with large genome sizes, is mostly distributed in freshwater and terrestrial environments. However, these bacteria have recently also been found in locations relevant to human health. Since the antimi- crobial resistance genes (AMR) from environmental resistome have the potential to be transferred to pathogens, it is essential to explore the resistant capabilities of environmental bacteria. In this study, the reconstruction of in silico resistome was performed for all nine available gemmata genomes. Furthermore, the genome of the newly isolated yet-undescribed strain G18 was sequenced and added to all analyses steps. Selected genomes were screened for the presence of mobile genetic elements. The flanking location of mobilizable genomic milieu around the AMR genes was of particular in- terest since such colocalization may appear to promote the horizontal gene transfer (HGT) events. Moreover the antibiotic susceptibility profile of six phylogenetically distinct strains of Gemmataceae planctomycetes was determined. Citation: Ivanova, A.A.; Keywords: planctomycetes; Gemmataceae; antibiotic resistance profile; resistome; mobilome Miroshnikov, K.K.; Oshkin, I.Y. Exploring Antibiotic Susceptibility, Resistome and Mobilome Structure of Planctomycetes from Gemmataceae 1. Introduction Family. Sustainability 2021, 13, 5031. Several decades ago humanity faced the global issue of growing antibiotic resistance https://doi.org/10.3390/su13095031 of bacterial pathogens in clinic [1–5]. -

Expanding the Chlamydiae Tree
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 2040 Expanding the Chlamydiae tree Insights into genome diversity and evolution JENNAH E. DHARAMSHI ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-513-1203-3 UPPSALA urn:nbn:se:uu:diva-439996 2021 Dissertation presented at Uppsala University to be publicly examined in A1:111a, Biomedical Centre (BMC), Husargatan 3, Uppsala, Tuesday, 8 June 2021 at 13:15 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Prof. Dr. Alexander Probst (Faculty of Chemistry, University of Duisburg-Essen). Abstract Dharamshi, J. E. 2021. Expanding the Chlamydiae tree. Insights into genome diversity and evolution. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 2040. 87 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-1203-3. Chlamydiae is a phylum of obligate intracellular bacteria. They have a conserved lifecycle and infect eukaryotic hosts, ranging from animals to amoeba. Chlamydiae includes pathogens, and is well-studied from a medical perspective. However, the vast majority of chlamydiae diversity exists in environmental samples as part of the uncultivated microbial majority. Exploration of microbial diversity in anoxic deep marine sediments revealed diverse chlamydiae with high relative abundances. Using genome-resolved metagenomics various marine sediment chlamydiae genomes were obtained, which significantly expanded genomic sampling of Chlamydiae diversity. These genomes formed several new clades in phylogenomic analyses, and included Chlamydiaceae relatives. Despite endosymbiosis-associated genomic features, hosts were not identified, suggesting chlamydiae with alternate lifestyles. Genomic investigation of Anoxychlamydiales, newly described here, uncovered genes for hydrogen metabolism and anaerobiosis, suggesting they engage in syntrophic interactions. -

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

Global Metagenomic Survey Reveals a New Bacterial Candidate Phylum in Geothermal Springs
ARTICLE Received 13 Aug 2015 | Accepted 7 Dec 2015 | Published 27 Jan 2016 DOI: 10.1038/ncomms10476 OPEN Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs Emiley A. Eloe-Fadrosh1, David Paez-Espino1, Jessica Jarett1, Peter F. Dunfield2, Brian P. Hedlund3, Anne E. Dekas4, Stephen E. Grasby5, Allyson L. Brady6, Hailiang Dong7, Brandon R. Briggs8, Wen-Jun Li9, Danielle Goudeau1, Rex Malmstrom1, Amrita Pati1, Jennifer Pett-Ridge4, Edward M. Rubin1,10, Tanja Woyke1, Nikos C. Kyrpides1 & Natalia N. Ivanova1 Analysis of the increasing wealth of metagenomic data collected from diverse environments can lead to the discovery of novel branches on the tree of life. Here we analyse 5.2 Tb of metagenomic data collected globally to discover a novel bacterial phylum (‘Candidatus Kryptonia’) found exclusively in high-temperature pH-neutral geothermal springs. This lineage had remained hidden as a taxonomic ‘blind spot’ because of mismatches in the primers commonly used for ribosomal gene surveys. Genome reconstruction from metagenomic data combined with single-cell genomics results in several high-quality genomes representing four genera from the new phylum. Metabolic reconstruction indicates a heterotrophic lifestyle with conspicuous nutritional deficiencies, suggesting the need for metabolic complementarity with other microbes. Co-occurrence patterns identifies a number of putative partners, including an uncultured Armatimonadetes lineage. The discovery of Kryptonia within previously studied geothermal springs underscores the importance of globally sampled metagenomic data in detection of microbial novelty, and highlights the extraordinary diversity of microbial life still awaiting discovery. 1 Department of Energy Joint Genome Institute, Walnut Creek, California 94598, USA. 2 Department of Biological Sciences, University of Calgary, Calgary, Alberta T2N 1N4, Canada. -

From Sporulation to Intracellular Offspring Production: Evolution
FROM SPORULATION TO INTRACELLULAR OFFSPRING PRODUCTION: EVOLUTION OF THE DEVELOPMENTAL PROGRAM OF EPULOPISCIUM A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by David Alan Miller January 2012 © 2012 David Alan Miller FROM SPORULATION TO INTRACELLULAR OFFSPRING PRODUCTION: EVOLUTION OF THE DEVELOPMENTAL PROGRAM OF EPULOPISCIUM David Alan Miller, Ph. D. Cornell University 2012 Epulopiscium sp. type B is an unusually large intestinal symbiont of the surgeonfish Naso tonganus. Unlike most other bacteria, Epulopiscium sp. type B has never been observed to undergo binary fission. Instead, to reproduce, it forms multiple intracellular offspring. We believe this process is related to endospore formation, an ancient and complex developmental process performed by certain members of the Firmicutes. Endospore formation has been studied for over 50 years and is best characterized in Bacillus subtilis. To study the evolution of endospore formation in the Firmicutes and the relatedness of this process to intracellular offspring formation in Epulopiscium, we have searched for sporulation genes from the B. subtilis model in all of the completed genomes of members of the Firmicutes, in addition to Epulopiscium sp. type B and its closest relative, the spore-forming Cellulosilyticum lentocellum. By determining the presence or absence of spore genes, we see the evolution of endospore formation in closely related bacteria within the Firmicutes and begin to predict if 19 previously characterized non-spore-formers have the genetic capacity to form a spore. We can also map out sporulation-specific mechanisms likely being used by Epulopiscium for offspring formation. -

Development of the Equine Hindgut Microbiome in Semi-Feral and Domestic Conventionally-Managed Foals Meredith K
Tavenner et al. Animal Microbiome (2020) 2:43 Animal Microbiome https://doi.org/10.1186/s42523-020-00060-6 RESEARCH ARTICLE Open Access Development of the equine hindgut microbiome in semi-feral and domestic conventionally-managed foals Meredith K. Tavenner1, Sue M. McDonnell2 and Amy S. Biddle1* Abstract Background: Early development of the gut microbiome is an essential part of neonate health in animals. It is unclear whether the acquisition of gut microbes is different between domesticated animals and their wild counterparts. In this study, fecal samples from ten domestic conventionally managed (DCM) Standardbred and ten semi-feral managed (SFM) Shetland-type pony foals and dams were compared using 16S rRNA sequencing to identify differences in the development of the foal hindgut microbiome related to time and management. Results: Gut microbiome diversity of dams was lower than foals overall and within groups, and foals from both groups at Week 1 had less diverse gut microbiomes than subsequent weeks. The core microbiomes of SFM dams and foals had more taxa overall, and greater numbers of taxa within species groups when compared to DCM dams and foals. The gut microbiomes of SFM foals demonstrated enhanced diversity of key groups: Verrucomicrobia (RFP12), Ruminococcaceae, Fusobacterium spp., and Bacteroides spp., based on age and management. Lactic acid bacteria Lactobacillus spp. and other Lactobacillaceae genera were enriched only in DCM foals, specifically during their second and third week of life. Predicted microbiome functions estimated computationally suggested that SFM foals had higher mean sequence counts for taxa contributing to the digestion of lipids, simple and complex carbohydrates, and protein. -

Card Uses a Minor Groove Wedge Mechanism to Stabilize the RNA
1 CarD uses a minor groove wedge mechanism to stabilize the RNA 2 polymerase open promoter complex 3 4 Brian Bae1, James Chen1, Elizabeth Davis1, Katherine Leon1, Seth A. Darst1,*, 5 Elizabeth A. Campbell1,* 6 7 1The Rockefeller University, Laboratory for Molecular Biophysics, 1230 York Avenue, New York, 8 NY 10065, USA. 9 10 *Correspondence to: E-mail: [email protected], [email protected] 11 12 Present Address: Elizabeth Davis, The University of Minnesota School of Medicine, 13 420 Delaware St. SE, Minneapolis, MN 55455, USA; Katherine Leon, Department of 14 Biochemistry and Molecular Biology, University of Chicago, 929 East 57th Street, GCIS 15 W219 Chicago, IL 60637, USA. 16 17 2 18 Abstract A key point to regulate gene expression is at transcription initiation, and 19 activators play a major role. CarD, an essential activator in Mycobacterium tuberculosis, 20 is found in many bacteria, including Thermus species, but absent in Escherichia coli. To 21 delineate the molecular mechanism of CarD, we determined crystal structures of 22 Thermus transcription initiation complexes containing CarD. The structures show CarD 23 interacts with the unique DNA topology presented by the upstream double- 24 stranded/single-stranded DNA junction of the transcription bubble. We confirm that our 25 structures correspond to functional activation complexes, and extend our understanding 26 of the role of a conserved CarD Trp residue that serves as a minor groove wedge, 27 preventing collapse of the transcription bubble to stabilize the transcription initiation 28 complex. Unlike E. coli RNAP, many bacterial RNAPs form unstable promoter 29 complexes, explaining the need for CarD. -

1 Strong Purifying Selection Is Associated with Genome
1 Strong Purifying Selection is Associated with Genome Streamlining in Epipelagic Marinimicrobia Carolina Alejandra Martinez-Gutierrez and Frank O. Aylward Department of Biological Sciences, Virginia Tech, Blacksburg, VA Email for correspondence: [email protected] Abstract Marine microorganisms inhabiting nutrient-depleted waters play critical roles in global biogeochemical cycles due to their abundance and broad distribution. Many of these microbes share similar genomic features including small genome size, low % G+C content, short intergenic regions, and low nitrogen content in encoded amino acid residue side chains (N-ARSC), but the evolutionary drivers of these characteristics are unclear. Here we compared the strength of purifying selection across the Marinimicrobia, a candidate phylum which encompasses a broad range of phylogenetic groups with disparate genomic features, by estimating the ratio of non-synonymous and synonymous substitutions (dN/dS) in conserved marker genes. Our analysis reveals that epipelagic Marinimicrobia that exhibit features consistent with genome streamlining have significantly lower dN/dS values when compared to the mesopelagic counterparts. We also found a significant positive correlation between median dN/dS values and % G+C content, N-ARSC, and intergenic region length. We did not identify a significant correlation between dN/dS ratios and estimated genome size, suggesting the strength of selection is not a primary factor shaping genome size in this group. Our findings are generally consistent with genome streamlining theory, which postulates that many genomic features of abundant epipelagic bacteria are the result of adaptation to oligotrophic nutrient conditions. Our results are also in agreement with previous findings that genome streamlining is common in epipelagic waters, suggesting that genomes inhabiting this region of the ocean have been shaped by strong selection together with prevalent nutritional constraints characteristic of this environment. -
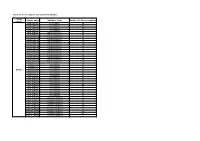
Table S1: List of Samples Included in the Analysis
Table S1: list of samples included in the analysis Study Sample name Inhibitory status Number of days at sampling number DNA.0P2T4 No inhibition 29 DNA.0P2T6 No inhibition 57 DNA.10P2T4 No inhibition 29 DNA.10P2T6 No inhibition 57 DNA.75P2T4 Phenol inhibition 29 DNA.75P2T6 Phenol inhibition 57 DNA.100P2T4 Phenol inhibition 29 DNA.100P2T6 Phenol inhibition 57 DNA.125P1T4 Phenol inhibition 29 DNA.125P1T6 Phenol inhibition 57 DNA.125P2T4 Phenol inhibition 29 DNA.125P2T6 Phenol inhibition 57 DNA.125P3T4 Phenol inhibition 29 DNA.125P3T6 Phenol inhibition 57 DNA.150P2T4 Phenol inhibition 29 DNA.150P2T6 Phenol inhibition 57 DNA.200P2T4 Phenol inhibition 29 DNA.200P2T6 Phenol inhibition 57 DNA.0N2T4 No inhibition 29 DNA.0N2T5 No inhibition 42 DNA.0N2T6 No inhibition 57 Study 1 DNA.5N2T4 No inhibition 29 DNA.5N2T5 No inhibition 42 DNA.5N2T6 No inhibition 57 DNA.10N2T4 No inhibition 29 DNA.10N2T5 No inhibition 42 DNA.10N2T6 No inhibition 57 DNA.15N2T4 No inhibition 29 DNA.15N2T5 No inhibition 42 DNA.15N2T6 No inhibition 57 DNA.25N2T4 No inhibition 29 DNA.25N2T5 No inhibition 42 DNA.25N2T6 No inhibition 57 DNA.75N2T4 Ammonia inhibition 29 DNA.75N2T5 Ammonia inhibition 42 DNA.75N2T6 Ammonia inhibition 57 DNA.100N2T4 Ammonia inhibition 29 DNA.100N2T5 Ammonia inhibition 42 DNA.100N2T6 Ammonia inhibition 57 DNA.250N2T4 Ammonia inhibition 29 DNA.250N2T5 Ammonia inhibition 42 DNA.250N2T6 Ammonia inhibition 57 nono2T3 No inhibition 16 noN2T4 Ammonia inhibition 23 noN2T8 Ammonia inhibition 60 noN2T9 Ammonia inhibition 85 noPhi2T4 Phenol inhibition 23 noPhi2T5 -

Characterization of the First Cultured Representative of Verrucomicrobia Subdivision 5 Indicates the Proposal of a Novel Phylum
The ISME Journal (2016) 10, 2801–2816 OPEN © 2016 International Society for Microbial Ecology All rights reserved 1751-7362/16 www.nature.com/ismej ORIGINAL ARTICLE Characterization of the first cultured representative of Verrucomicrobia subdivision 5 indicates the proposal of a novel phylum Stefan Spring1, Boyke Bunk2, Cathrin Spröer3, Peter Schumann3, Manfred Rohde4, Brian J Tindall1 and Hans-Peter Klenk1,5 1Department Microorganisms, Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany; 2Department Microbial Ecology and Diversity Research, Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany; 3Department Central Services, Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany and 4Central Facility for Microscopy, Helmholtz-Centre of Infection Research, Braunschweig, Germany The recently isolated strain L21-Fru-ABT represents moderately halophilic, obligately anaerobic and saccharolytic bacteria that thrive in the suboxic transition zones of hypersaline microbial mats. Phylogenetic analyses based on 16S rRNA genes, RpoB proteins and gene content indicated that strain L21-Fru-ABT represents a novel species and genus affiliated with a distinct phylum-level lineage originally designated Verrucomicrobia subdivision 5. A survey of environmental 16S rRNA gene sequences revealed that members of this newly recognized phylum are wide-spread and ecologically important in various anoxic environments ranging from hypersaline sediments to wastewater and the intestine of animals. Characteristic phenotypic traits of the novel strain included the formation of extracellular polymeric substances, a Gram-negative cell wall containing peptidoglycan and the absence of odd-numbered cellular fatty acids. Unusual metabolic features deduced from analysis of the genome sequence were the production of sucrose as osmoprotectant, an atypical glycolytic pathway lacking pyruvate kinase and the synthesis of isoprenoids via mevalonate. -

Molecular Diversity and Ecology of Microbial Plankton Stephen J
02 Giovanni 9-14 6/9/05 9:22 AM Page 9 NATURE|Vol 437|15 September 2005|doi:10.1038/nature04158 INSIGHT REVIEW Molecular diversity and ecology of microbial plankton Stephen J. Giovannoni1 & Ulrich Stingl1 The history of microbial evolution in the oceans is probably as old as the history of life itself. In contrast to terrestrial ecosystems, microorganisms are the main form of biomass in the oceans, and form some of the largest populations on the planet. Theory predicts that selection should act more efficiently in large populations. But whether microbial plankton populations harbour organisms that are models of adaptive sophistication remains to be seen. Genome sequence data are piling up, but most of the key microbial plankton clades have no cultivated representatives, and information about their ecological activities is sparse. Certain characteristics of the ocean environment — the prevailing cultivation of key organisms, metagenomics and ongoing biogeo- low-nutrient state of the ocean surface, in particular — mean it is chemical studies. It seems very likely that the biology of the dominant sometimes regarded as an extreme ecosystem. Fixed forms of nitrogen, microbial plankton groups will be unravelled in the years ahead. phosphorus and iron are often at very low or undetectable levels in the Here we review current knowledge about marine bacterial and ocean’s circulatory gyres, which occur in about 70% of the oceans1. archaeal diversity, as inferred from phylogenies of genes recovered Photosynthesis is the main source of metabolic energy and the basis of from the ocean water column, and consider the implications of micro- the food chain; ocean phytoplankton account for nearly 50% of global bial diversity for understanding the ecology of the oceans. -

Diversity and Community Structure of Marine Microbes Around the Benham Rise Underwater Plateau, Northeastern Philippines
Diversity and community structure of marine microbes around the Benham Rise underwater plateau, northeastern Philippines Andrian P. Gajigan1,2, Aletta T. Yñiguez1, Cesar L. Villanoy1, Maria Lourdes San Diego-McGlone1, Gil S. Jacinto1 and Cecilia Conaco1 1 Marine Science Institute, University of the Philippines Diliman, Quezon City, Philippines 2 Current affiliation: Department of Oceanography, University of Hawaii at Manoa, USA ABSTRACT Microbes are central to the structuring and functioning of marine ecosystems. Given the remarkable diversity of the ocean microbiome, uncovering marine microbial taxa remains a fundamental challenge in microbial ecology. However, there has been little effort, thus far, to describe the diversity of marine microorganisms in the region of high marine biodiversity around the Philippines. Here, we present data on the taxonomic diversity of bacteria and archaea in Benham Rise, Philippines, Western Pacific Ocean, using 16S V4 rRNA gene sequencing. The major bacterial and archaeal phyla identified in the Benham Rise are Proteobacteria, Cyanobacteria, Actinobacteria, Bacteroidetes, Marinimicrobia, Thaumarchaeota and, Euryarchaeota. The upper mesopelagic layer exhibited greater microbial diversity and richness compared to surface waters. Vertical zonation of the microbial community is evident and may be attributed to physical stratification of the water column acting as a dispersal barrier. Canonical Corre- spondence Analysis (CCA) recapitulated previously known associations of taxa and physicochemical parameters in the environment, such as the association of oligotrophic clades with low nutrient surface water and deep water clades that have the capacity to oxidize ammonia or nitrite at the upper mesopelagic layer. These findings provide Submitted 8 January 2018 foundational information on the diversity of marine microbes in Philippine waters.