Gene Conservation Among Endospore-Forming Bacteria Reveals Additional
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

From Sporulation to Intracellular Offspring Production: Evolution
FROM SPORULATION TO INTRACELLULAR OFFSPRING PRODUCTION: EVOLUTION OF THE DEVELOPMENTAL PROGRAM OF EPULOPISCIUM A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by David Alan Miller January 2012 © 2012 David Alan Miller FROM SPORULATION TO INTRACELLULAR OFFSPRING PRODUCTION: EVOLUTION OF THE DEVELOPMENTAL PROGRAM OF EPULOPISCIUM David Alan Miller, Ph. D. Cornell University 2012 Epulopiscium sp. type B is an unusually large intestinal symbiont of the surgeonfish Naso tonganus. Unlike most other bacteria, Epulopiscium sp. type B has never been observed to undergo binary fission. Instead, to reproduce, it forms multiple intracellular offspring. We believe this process is related to endospore formation, an ancient and complex developmental process performed by certain members of the Firmicutes. Endospore formation has been studied for over 50 years and is best characterized in Bacillus subtilis. To study the evolution of endospore formation in the Firmicutes and the relatedness of this process to intracellular offspring formation in Epulopiscium, we have searched for sporulation genes from the B. subtilis model in all of the completed genomes of members of the Firmicutes, in addition to Epulopiscium sp. type B and its closest relative, the spore-forming Cellulosilyticum lentocellum. By determining the presence or absence of spore genes, we see the evolution of endospore formation in closely related bacteria within the Firmicutes and begin to predict if 19 previously characterized non-spore-formers have the genetic capacity to form a spore. We can also map out sporulation-specific mechanisms likely being used by Epulopiscium for offspring formation. -
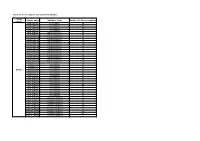
Table S1: List of Samples Included in the Analysis
Table S1: list of samples included in the analysis Study Sample name Inhibitory status Number of days at sampling number DNA.0P2T4 No inhibition 29 DNA.0P2T6 No inhibition 57 DNA.10P2T4 No inhibition 29 DNA.10P2T6 No inhibition 57 DNA.75P2T4 Phenol inhibition 29 DNA.75P2T6 Phenol inhibition 57 DNA.100P2T4 Phenol inhibition 29 DNA.100P2T6 Phenol inhibition 57 DNA.125P1T4 Phenol inhibition 29 DNA.125P1T6 Phenol inhibition 57 DNA.125P2T4 Phenol inhibition 29 DNA.125P2T6 Phenol inhibition 57 DNA.125P3T4 Phenol inhibition 29 DNA.125P3T6 Phenol inhibition 57 DNA.150P2T4 Phenol inhibition 29 DNA.150P2T6 Phenol inhibition 57 DNA.200P2T4 Phenol inhibition 29 DNA.200P2T6 Phenol inhibition 57 DNA.0N2T4 No inhibition 29 DNA.0N2T5 No inhibition 42 DNA.0N2T6 No inhibition 57 Study 1 DNA.5N2T4 No inhibition 29 DNA.5N2T5 No inhibition 42 DNA.5N2T6 No inhibition 57 DNA.10N2T4 No inhibition 29 DNA.10N2T5 No inhibition 42 DNA.10N2T6 No inhibition 57 DNA.15N2T4 No inhibition 29 DNA.15N2T5 No inhibition 42 DNA.15N2T6 No inhibition 57 DNA.25N2T4 No inhibition 29 DNA.25N2T5 No inhibition 42 DNA.25N2T6 No inhibition 57 DNA.75N2T4 Ammonia inhibition 29 DNA.75N2T5 Ammonia inhibition 42 DNA.75N2T6 Ammonia inhibition 57 DNA.100N2T4 Ammonia inhibition 29 DNA.100N2T5 Ammonia inhibition 42 DNA.100N2T6 Ammonia inhibition 57 DNA.250N2T4 Ammonia inhibition 29 DNA.250N2T5 Ammonia inhibition 42 DNA.250N2T6 Ammonia inhibition 57 nono2T3 No inhibition 16 noN2T4 Ammonia inhibition 23 noN2T8 Ammonia inhibition 60 noN2T9 Ammonia inhibition 85 noPhi2T4 Phenol inhibition 23 noPhi2T5 -

Gene Conservation Among Endospore-Forming Bacteria Reveals Additional Sporulation Genes in Bacillus Subtilis
Gene Conservation among Endospore-Forming Bacteria Reveals Additional Sporulation Genes in Bacillus subtilis Bjorn A. Traag,a Antonia Pugliese,a Jonathan A. Eisen,b Richard Losicka Department of Molecular and Cellular Biology, Harvard University, Cambridge, Massachusetts, USAa; Department of Evolution and Ecology, University of California Davis Genome Center, Davis, California, USAb The capacity to form endospores is unique to certain members of the low-G؉C group of Gram-positive bacteria (Firmicutes) and requires signature sporulation genes that are highly conserved across members of distantly related genera, such as Clostridium and Bacillus. Using gene conservation among endospore-forming bacteria, we identified eight previously uncharacterized genes that are enriched among endospore-forming species. The expression of five of these genes was dependent on sporulation-specific transcription factors. Mutants of none of the genes exhibited a conspicuous defect in sporulation, but mutants of two, ylxY and ylyA, were outcompeted by a wild-type strain under sporulation-inducing conditions, but not during growth. In contrast, a ylmC mutant displayed a slight competitive advantage over the wild type specific to sporulation-inducing conditions. The phenotype of a ylyA mutant was ascribed to a defect in spore germination efficiency. This work demonstrates the power of combining phy- logenetic profiling with reverse genetics and gene-regulatory studies to identify unrecognized genes that contribute to a con- served developmental process. he formation of endospores is a distinctive developmental the successive actions of four compartment-specific sigma factors Tprocess wherein a dormant cell type (the endospore) is formed (appearing in the order F, E, G, and K), whose activities are inside another cell (the mother cell) and ultimately released into confined to the forespore (F and G) or the mother cell (E and the environment by lysis of the mother cell (1, 2). -

A Metagenomic Window Into the 2-Km-Deep Terrestrial Subsurface Aquifer Revealed Multiple Pathways of Organic Matter Decomposition Vitaly V
FEMS Microbiology Ecology, 94, 2018, fiy152 doi: 10.1093/femsec/fiy152 Advance Access Publication Date: 7 August 2018 Research Article RESEARCH ARTICLE A metagenomic window into the 2-km-deep terrestrial subsurface aquifer revealed multiple pathways of organic matter decomposition Vitaly V. Kadnikov1, Andrey V. Mardanov1, Alexey V. Beletsky1, David Banks3, Nikolay V. Pimenov4, Yulia A. Frank2,OlgaV.Karnachuk2 and Nikolai V. Ravin1,* 1Institute of Bioengineering, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky prosp. 33-2, Moscow, 119071, Russia, 2Laboratory of Biochemistry and Molecular Biology, Tomsk State University, Lenina prosp. 35, Tomsk, 634050, Russia, 3School of Engineering, Systems Power & Energy, Glasgow University, Glasgow G12 8QQ, and Holymoor Consultancy Ltd., 360 Ashgate Road, Chesterfield, Derbyshire S40 4BW, UK and 4Winogradsky Institute of Microbiology, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky prosp 33-2, Moscow, 119071, Russia ∗Corresponding author: Institute of Bioengineering, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky prosp. 33-2, Moscow, 119071, Russia. Tel: +74997833264; Fax: +74991353051; E-mail: [email protected] One sentence summary: metagenome-assembled genomes of microorganisms from the deep subsurface thermal aquifer revealed functional diversity of the community and novel uncultured bacterial lineages Editor: Tillmann Lueders ABSTRACT We have sequenced metagenome of the microbial community of a deep subsurface thermal aquifer in the Tomsk Region of the Western Siberia, Russia. Our goal was the recovery of near-complete genomes of the community members to enable accurate reconstruction of metabolism and ecological roles of the microbial majority, including previously unstudied lineages. The water, obtained via a 2.6 km deep borehole 1-R, was anoxic, with a slightly alkaline pH, and a temperature around 45◦C. -

Analysis of Microbial Community Structure of Pit Mud for Chinese Strong-Flavor Liquor Fermentation Using Next Generation DNA Sequencing of Full-Length 16S Rrna
bioRxiv preprint doi: https://doi.org/10.1101/380949; this version posted July 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Analysis of microbial community structure of pit mud for Chinese strong-flavor liquor fermentation using next generation DNA sequencing of full-length 16S rRNA Zuolin Liu1†, Ying Han1†, Junwei Li1,2, Runjie Cao2, Hongkui He2*, Anjun Li2 and Zhizhou Zhang1* 1School of Chemical Engineering and Chemistry, Harbin Institute of Technology, Harbin, China 150006 2The Center for Solid-state Fermentation Engineering of Anhui Province, The Anhui GujingTribute Liquor Ltd, Bozhou, Anhui, China, 236800 † *The corresponding author: [email protected]; [email protected] Equal contribution Contact: Zhizhou Zhang, PhD Prof [email protected] 86-631-5683176 Abstract. The pit is the necessary bioreactor for brewing process of Chinese strong-flavor liquor. Pit mud in pits contains a large number of microorganisms and is a complex ecosystem. The analysis of bacterial flora in pit mud is of great significance to understand liquor fermentation mechanisms. To overcome taxonomic limitations of short reads in 16S rRNA variable region sequencing, we used high-throughput DNA sequencing of near full-length 16S rRNA gene to analyze microbial compositions of different types of pit mud that produce different qualities of strong-flavor liquor. The results showed that the main species in pit mud were Pseudomonas extremaustralis 14-3, Pseudomonas veronii, Serratia marcescens WW4, and Clostridium leptum in Ruminiclostridium. -

The Core Populations and Co-Occurrence Patterns Of
Rui et al. Biotechnol Biofuels (2015) 8:158 DOI 10.1186/s13068-015-0339-3 RESEARCH Open Access The core populations and co‑occurrence patterns of prokaryotic communities in household biogas digesters Junpeng Rui1,2†, Jiabao Li1,2†, Shiheng Zhang1,2, Xuefeng Yan1,2, Yuanpeng Wang3 and Xiangzhen Li1,2* Abstract Background: Household biogas digesters are widely used to harvest energy in rural areas of developing countries. Understanding core prokaryotic communities, their co-occurrence patterns, and their relationships to environmental factors is important to manage these small-scale anaerobic digestion systems effectively. In this study, 43 household biogas digesters were collected across eight provinces in China. Prokaryotic communities were investigated using 454 pyrosequencing of 16S rRNA genes. Results: Fourteen core genera and ten core OTUs were identified in household biogas digesters. They were mainly affiliated with the phylum Firmicutes, Synergistetes, Actinobacteria, Chloroflexi, and Spirochaetes. Core prokaryotic genera were mainly composed of Clostridium, Clostridium XI, Syntrophomonas, Cloacibacillus, Sedimentibacter, and Turicibacter. Prokaryotic communities in the 43 samples were clearly divided into two clusters. Cluster I was domi- nated by Clostridium, while Cluster II was dominated by members of Spirochaetes, Bacteroidales, Clostridia, and abundant syntrophs and methanogens. NH4+-N and COD contributed significantly to the assembly of the prokaryotic community in Cluster I, while NH4+-N, pH, and phosphate contributed significantly to Cluster II. Correlation-based network analysis showed that the prokaryotic communities in the biogas digesters were dominated by some func- tional modules. Cluster I was dominated by acetotrophic methanogenic modules and the Clostridium-driven primary fermentation module, while the network of Cluster II was dominated by hydrogenotrophic and acetogenic methano- genesis modules and multi-group-driven (Spirochaetes, Bacteroidales, and Clostridia) primary fermentation modules. -

DNA-SIP Identifies Sulfate-Reducing Clostridia As Important Toluene Degraders in Tar-Oil-Contaminated Aquifer Sediment
The ISME Journal (2010) 4, 1314–1325 & 2010 International Society for Microbial Ecology All rights reserved 1751-7362/10 www.nature.com/ismej ORIGINAL ARTICLE DNA-SIP identifies sulfate-reducing Clostridia as important toluene degraders in tar-oil-contaminated aquifer sediment Christian Winderl, Holger Penning, Frederick von Netzer, Rainer U Meckenstock and Tillmann Lueders Institute of Groundwater Ecology, Helmholtz Zentrum Mu¨nchen—German Research Centre for Environmental Health, Neuherberg, Germany Global groundwater resources are constantly challenged by a multitude of contaminants such as aromatic hydrocarbons. Especially in anaerobic habitats, a large diversity of unrecognized microbial populations may be responsible for their degradation. Still, our present understanding of the respective microbiota and their ecophysiology is almost exclusively based on a small number of cultured organisms, mostly within the Proteobacteria. Here, by DNA-based stable isotope probing (SIP), we directly identified the most active sulfate-reducing toluene degraders in a diverse sedimentary microbial community originating from a tar-oil-contaminated aquifer at a former coal gasification plant. 13 13 On incubation of fresh sediments with C7-toluene, the production of both sulfide and CO2 was clearly coupled to the 13C-labeling of DNA of microbes related to Desulfosporosinus spp. within the Peptococcaceae (Clostridia). The screening of labeled DNA fractions also suggested a novel benzylsuccinate synthase alpha-subunit (bssA) sequence type previously only detected in the environment to be tentatively affiliated with these degraders. However, carbon flow from the contaminant into degrader DNA was only B50%, pointing toward high ratios of heterotrophic CO2-fixation during assimilation of acetyl-CoA originating from the contaminant by these degraders. -

Nutritional Features of Syntrophomonas Wolfei P
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Oct. 1990, p. 3223-3224 Vol. 56, No. 10 0099-2240/90/103223-02$02.00/0 Copyright © 1990, American Society for Microbiology Nutritional Features of Syntrophomonas wolfei P. SHAWN BEATY AND MICHAEL J. McINERNEY* Department ofBotany and Microbiology, University of Oklahoma, Norman, Oklahoma 73019-0245 Received 27 March 1990/Accepted 18 July 1990 Syntrophomonas wolfei subsp. wolfei grew poorly in a defined medium with crotonate as the energy source in the absence of rumen fluid. Thiamine, lipoic acid, biotin, cyanocobalamin, and para-aminobenzoic acid were required for growth comparable to that obtained with the rumen fluid-based medium. Iron and cobalt were also required for the growth of S. wolfei in the chemically defined medium. Propionate and longer-chain fatty acids are important in medium lacking a particular compound before it was intermediates in the conversion of organic matter to CH4 and concluded that the compound was not required for the CO2 (4). These compounds are degraded to methanogenic growth. Growth was monitored spectrophotometrically at substrates (acetate, H2, and formate) by syntrophic bacteria 600 nm, and growth rates were calculated from changes in which require the presence of methanogens or other H2- absorbance (6). using bacteria to maintain low concentrations of H2 for the The pure culture of S. wolfei grew at a specific growth rate degradation offatty acids to be thermodynamically favorable of 0.039/h in crotonate basal medium which contained rumen (4). Syntrophomonas wolfei subsp. wolfei is an anaerobic fluid. Decreasing the rumen fluid concentration of this me- bacterium that a-oxidizes short-chain saturated fatty acids, dium from 5 to 0.5% (vol/vol) did not affect the rate of four to eight carbons in length, to acetate, propionate (from growth or the final absorbance (0.45 to 0.5) of the culture odd-numbered fatty acids), and H2 only when grown in (data not shown). -

Research Article Archaea and Bacteria Acclimate to High Total Ammonia in a Methanogenic Reactor Treating Swine Waste
Hindawi Publishing Corporation Archaea Volume 2016, Article ID 4089684, 10 pages http://dx.doi.org/10.1155/2016/4089684 Research Article Archaea and Bacteria Acclimate to High Total Ammonia in a Methanogenic Reactor Treating Swine Waste Sofia Esquivel-Elizondo,1,2 Prathap Parameswaran,3 Anca G. Delgado,1 Juan Maldonado,1 Bruce E. Rittmann,1,2 and Rosa Krajmalnik-Brown1,2 1 Swette Center for Environmental Biotechnology, The Biodesign Institute, Arizona State University, P.O. Box 875701, Tempe, AZ 85287-5701, USA 2School of Sustainable Engineering and the Built Environment, Arizona State University, Tempe, AZ, USA 3Department of Civil Engineering, Kansas State University, 2118 Fiedler Hall, Manhattan, KS 66506, USA Correspondence should be addressed to Rosa Krajmalnik-Brown; [email protected] Received 10 June 2016; Accepted 11 August 2016 Academic Editor: Jessica A. Smith Copyright © 2016 Sofia Esquivel-Elizondo et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Inhibition by ammonium at concentrations above 1000 mgN/L is known to harm the methanogenesis phase of anaerobic digestion. We anaerobically digested swine waste and achieved steady state COD-removal efficiency of around 52% with no fatty-acid or H2 accumulation. As the anaerobic microbial community adapted to the gradual increase of total ammonia-N (NH3-N) from 890 ± 295 to 2040 ± 30 mg/L, the Bacterial and Archaeal communities became less diverse. Phylotypes most closely related to hydrogenotrophic Methanoculleus (36.4%) and Methanobrevibacter (11.6%), along with acetoclastic Methanosaeta (29.3%), became the most abundant Archaeal sequences during acclimation. -

Ecophysiology of Sulfate-Reducing Bacteria and Syntrophic Communities in Marine Anoxic Sediments” Derya Özüölmez Wageningen, 12 September 2017
Propositions 1. Prolonged incubation is a key strategy toward the enrichment of marine syntrophs. (this thesis) 2. Hydrogen-consuming methanogens can effectively compete with hydrogen- consuming sulfate reducers even at high sulfate concentration. (this thesis) 3. The plastic-eating wax worm, Galleria mellonella, can help cleaning up existing plastic mass (Bombelli et al. 2017, Current Biology 27(8): 292-293), but the real solution to environmental pollution still relies on shifting from disposable plastics to reusable and biodegradable materials. 4. With the discovery of abundant molecular hydrogen in Enceladus’ salty ocean (Waite et al. (2017) Science 356:6334, 155-159) extraterrestrial life in our solar system has become highly probable, and should shift our life-hunting focus from distant stars to our nearby neighbors. 5. With the "Help me to do it myself" motto, the Montessori education leads children to grow into mature, creative and self-confident adults. 6. Nothing is impossible as long as the dream is held close to the heart and patience and persistence are applied for the accomplishment. Propositions belonging to the PhD thesis entitled: “Ecophysiology of sulfate-reducing bacteria and syntrophic communities in marine anoxic sediments” Derya Özüölmez Wageningen, 12 September 2017 Ecophysiology of sulfate-reducing bacteria and syntrophic communities in marine anoxic sediments Derya Özüölmez Thesis committee Promotor Prof. Dr Alfons J.M. Stams Personal chair at the Laboratory of Microbiology Wageningen University & Research Co-promotor Dr Caroline M. Plugge Associate professor, Laboratory of Microbiology Wageningen University & Research Other members Prof. Dr Tinka Murk, Wageningen University & Research Prof. Dr Gerard Muyzer, University of Amsterdam, The Netherlands Prof. -

An Ensemble Approach to the Taxonomic Classification of Metatranscriptomic Sequence Data
bioRxiv preprint doi: https://doi.org/10.1101/081026; this version posted October 28, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Gist – an ensemble approach to the taxonomic classification of metatranscriptomic sequence data. Samantha Halliday1,2 and John Parkinson1,3,4,* 1 Program in Molecular Structure and Function, Hospital for Sick Children, Toronto, Ontario, M5G 1L7, Canada 2 Department of Computer Science, University of Toronto, Toronto, Ontario, M5S 1A8, Canada 3 Department of Molecular Genetics, University of Toronto, Toronto, Ontario, M5S 1A8, Canada 4 Department of Biochemistry, University of Toronto, Toronto, Ontario, M5S 1A8, Canada * To whom correspondence should be addressed: [email protected] Running Title: Taxonomic classification of metatranscriptomic data Keywords: metatranscriptomics, taxonomic classification, 1 bioRxiv preprint doi: https://doi.org/10.1101/081026; this version posted October 28, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT The study of whole microbial communities through RNA-seq, or metatranscriptomics, offers a unique view of the relative levels of activity for different genes across a large number of species simultaneously. To make sense of these sequencing data, it is necessary to be able to as- sign both taxonomic and functional identities to each sequenced read. High-quality identifica- tions are important not only for community profiling, but to also ensure that functional assign- ments of sequence reads are correctly attributed to their source taxa.