Acetylglutamate Kinase (EC 2.7.2.8)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METACYC ID Description A0AR23 GO:0004842 (Ubiquitin-Protein Ligase
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Heat Stress Responsive Zostera marina Genes, Southern Population (α=0. -

Mapping Active Site Residues in Glutamate-5-Kinase. the Substrate Glutamate and the Feed-Back Inhibitor Proline Bind at Overlapping Sites
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 580 (2006) 6247–6253 Mapping active site residues in glutamate-5-kinase. The substrate glutamate and the feed-back inhibitor proline bind at overlapping sites Isabel Pe´rez-Arellanoa, Vicente Rubiob, Javier Cerveraa,* a Centro de Investigacio´n Prı´ncipe Felipe, Avda. Autopista del Saler, 16, Valencia 46013, Spain b Instituto de Biomedicina de Valencia (IBV-CSIC), Jaime Roig 11, Valencia 46010, Spain Received 22 August 2006; revised 10 October 2006; accepted 12 October 2006 Available online 20 October 2006 Edited by Stuart Ferguson (a domain named after pseudo uridine synthases and archaeo- Abstract Glutamate-5-kinase (G5K) catalyzes the controlling first step of proline biosynthesis. Substrate binding, catalysis sine-specific transglycosylases) domain [10]. AAK domains and feed-back inhibition by proline are functions of the N-termi- bind ATP and a phosphorylatable acidic substrate, and make nal 260-residue domain of G5K. We study here the impact on a phosphoric anhydride [11]. The function of the PUA domain these functions of 14 site-directed mutations affecting 9 residues (a domain characteristically found in some RNA-modifying of Escherichia coli G5K, chosen on the basis of the structure of enzymes) [12] is not known. By deleting the PUA domain of the bisubstrate complex of the homologous enzyme acetylgluta- E. coli G5K we showed [10] that the isolated AAK domain, mate kinase (NAGK). The results support the predicted roles as the complete enzyme, forms tetramers, catalyzes the reac- of K10, K217 and T169 in catalysis and ATP binding and of tion and is feed-back inhibited by proline. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -
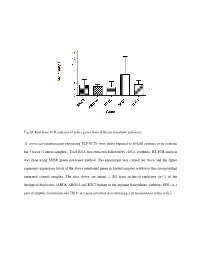
Fig.S1 Real-Time PCR Analysis of Select Genes from Different Metabolic Pathways S. Cerevisiae Transformants Expressing TEF-YCT1
Fig.S1 Real-time PCR analysis of select genes from different metabolic pathways S. cerevisiae transformants expressing TEF-YCT1 were either exposed to 500µM cysteine or no cysteine for 5 hours (Control samples). Total RNA was extracted followed by cDNA synthesis. RT-PCR analysis was done using SYBR green dye-based method. The experiment was carried out twice and the figure represents expression levels of the above mentioned genes in treated samples relative to the corresponding untreated control samples. The data above are means ± SD from technical replicates (n=3) of the biological duplicates. [ARG8, ARG5,6 and RTC2 belong to the arginine biosynthetic pathway, SSU1 is a part of sulphur metabolism and TIS11 is a gene involved in maintaining iron homeostasis in the cells.] Table S1: List of strains used in this study Strain Genotype Source ABC 1738 (yct1) MATα his31 leu20 lys20 ura30 YLL055W:: kanMX4 Euroscarf (Y01543) ABC3612(arg3) MATa his31 leu20 ura30 YJL088w::kanMX4 Euroscarf (Y11335) ABC4812(arg5,6) MATa his31 leu20 ura30 YER069w::kanMX4 Euroscarf (Y10209) ABC4811(arg8) MATa his31 leu20 ura30 YOL140w::kanMX4 Euroscarf (Y17711) ABC4814(spe1) MATa his31 leu20 ura30 YKL184w::kanMX4 Euroscarf (Y15034) ABC4815(spe2) MATa his31 leu20 ura30 YOL052c::kanMX4 Euroscarf (Y11743) ABC4816(spe3) MATa his31 leu20 ura30 YPR069c::kanMX4 Euroscarf (Y15488) ABC4817(spe4) MATa his31 leu20 ura30 YLR146c::kanMX4 Euroscarf (Y16945) ABC3604(ssu1) MATa his31 leu20 ura30 YPL092w::kanMX4 Euroscarf (Y12160) ABC1486(str2) MATa his31 leu20 ura30 YJR130c::kanMX4 -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

Saccharomyces Cerevisiae Aspartate Kinase Mechanism and Inhibition
In compliance with the Canadian Privacy Legislation some supporting forms may have been removed from this dissertation. While these forms may be included in the document page count, their removal does not represent any loss of content from the dissertation. Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry FUNGAL ASPARTATE KINASE MECHANISM AND INHIBITION By DAVID C. BAREICH, B.Sc. A Thesis Submitted to the School of Graduate Studies in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy McMaster University © Copyright by David C. Bareich, June 2003 1 Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry FUNGAL ASPARTATE KINASE MECHANISM AND INHIBITION Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry DOCTOR OF PHILOSOPHY (2003) McMaster University (Biochemistry) Hamilton, Ontario TITLE: Saccharomyces cerevisiae aspartate kinase mechanism and inhibition AUTHOR: David Christopher Bareich B.Sc. (University of Waterloo) SUPERVISOR: Professor Gerard D. Wright NUMBER OF PAGES: xix, 181 11 Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry ABSTRACT Aspartate kinase (AK) from Saccharomyces cerevisiae (AKsc) catalyzes the first step in the aspartate pathway responsible for biosynthesis of L-threonine, L-isoleucine, and L-methionine in fungi. Little was known about amino acids important for AKsc substrate binding and catalysis. Hypotheses about important amino acids were tested using site directed mutagenesis to substitute these amino acids with others having different properties. Steady state kinetic parameters and pH titrations of the variant enzymes showed AKsc-K18 and H292 to be important for binding and catalysis. Little was known about how the S. cerevisiae aspartate pathway kinases, AKsc and homoserine kinase (HSKsc), catalyze the transfer of the y-phosphate from adenosine triphosphate (ATP) to L-aspartate or L-homoserine, respectively. -

A NOVEL TWO-DOMAIN ARCHITECTURE WITHIN the AMINO ACID KINASE ENZYME FAMILY REVEALED by the CRYSTAL STRUCTURE of Escherichia Coli
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Digital.CSIC A NOVEL TWO-DOMAIN ARCHITECTURE WITHIN THE AMINO ACID KINASE ENZYME FAMILY REVEALED BY THE CRYSTAL STRUCTURE OF Escherichia coli GLUTAMATE 5-KINASE Clara Marco-Marín1, Fernando Gil-Ortiz,1 Isabel Pérez-Arellano,2 Javier Cervera,2 Ignacio Fita3 and Vicente Rubio1,* 1Instituto de Biomedicina de Valencia (IBV-CSIC) and Center for Biomedical Research on Rare Diseases (CIBERER-ISCIII), Jaume Roig 11, Valencia-46010, Spain 2Centro de Investigación Príncipe Felipe (FVIB-CSIC), Avda. Autopista del Saler 16, Valencia-46013, Spain 3Instituto de Biología Molecular de Barcelona (IBMB-CSIC). Institute for Research in Biomedicine. Parc Científic, Josep Samitier 1-5, 08028-Barcelona, Spain. Present address: F. Gil-Ortiz, Centro de Investigación Príncipe Felipe (FVIB-CSIC), Avda. Autopista del Saler 16, Valencia-46013, Spain * Corresponding author: Vicente Rubio Instituto de Biomedicina de Valencia Jaume Roig 11, Valencia-46010, Spain E-mail: [email protected] Tel. +34 963 391 772 Fax. +34 963 690 800 Short title: Structure of -glutamyl kinase of Escherichia coli 1 Summary. Glutamate 5-kinase (G5K) makes the highly unstable product glutamyl-5- phosphate (G5P) in the initial, controlling step of proline/ornithine synthesis, being feed-back inhibited by proline or ornithine, and causing, when defective, clinical hyperammonaemia. We have determined two crystal structures of G5K from Escherichia coli, at 2.9- and 2.5-Å-resolution, complexed with glutamate and sulphate, or with G5P, sulphate and the proline analog 5-oxoproline. E. coli G5K presents a novel tetrameric (dimer of dimers) architecture. -
Generate Metabolic Map Poster
Authors: Zheng Zhao, Delft University of Technology Marcel A. van den Broek, Delft University of Technology S. Aljoscha Wahl, Delft University of Technology Wilbert H. Heijne, DSM Biotechnology Center Roel A. Bovenberg, DSM Biotechnology Center Joseph J. Heijnen, Delft University of Technology An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Marco A. van den Berg, DSM Biotechnology Center Peter J.T. Verheijen, Delft University of Technology Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. PchrCyc: Penicillium rubens Wisconsin 54-1255 Cellular Overview Connections between pathways are omitted for legibility. Liang Wu, DSM Biotechnology Center Walter M. van Gulik, Delft University of Technology L-quinate phosphate a sugar a sugar a sugar a sugar multidrug multidrug a dicarboxylate phosphate a proteinogenic 2+ 2+ + met met nicotinate Mg Mg a cation a cation K + L-fucose L-fucose L-quinate L-quinate L-quinate ammonium UDP ammonium ammonium H O pro met amino acid a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar K oxaloacetate L-carnitine L-carnitine L-carnitine 2 phosphate quinic acid brain-specific hypothetical hypothetical hypothetical hypothetical -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -

Identification, Cloning and Expression of the Mouse N-Acetylglutamate Synthase Gene
Biochem. J. (2002) 364, 825–831 (Printed in Great Britain) 825 Identification, cloning and expression of the mouse N-acetylglutamate synthase gene Ljubica CALDOVIC*, Hiroki MORIZONO*, Xiaolin YU*, Mark THOMPSON*, Dashuang SHI*, Rene GALLEGOS†, Norma M. ALLEWELL‡, Michael H. MALAMY† and Mendel TUCHMAN*1 *Children’s Research Institute, Children’s National Medical Center, George Washington University, 111 Michigan Ave NW, Washington, DC 20010, U.S.A., †Department of Microbiology, Tufts University, 136 Harrison Ave, Boston, MA 02111, U.S.A., and ‡College of Life Sciences, University of Maryland, College Park, MD 20742, U.S.A. In ureotelic animals, N-acetylglutamate (NAG) is an essential expression levels in kidney, spleen and testis. The deduced amino allosteric activator of carbamylphosphate synthetase I (CPSI), acid sequence contains a putative mitochondrial targeting signal the first enzyme in the urea cycle. NAG synthase (NAGS; EC at the N-terminus. The cDNA sequence complements an argA 2.3.1.1) catalyses the formation of NAG from glutamate and (NAGS)-deficient Escherichia coli strain, reversing its arginine acetyl-CoA in liver and intestinal mitochondria. This enzyme is auxotrophy. His-tagged versions of the pre-protein and two supposed to regulate ureagenesis by producing variable amounts putative mature proteins were each overexpressed in E. coli, and of NAG, thus modulating CPSI activity. Moreover, inherited purified to apparent homogeneity by using a nickel-affinity deficiencies in NAGS have been associated with hyperammo- column. The pre-protein and the two putative mature proteins naemia, probably due to the loss of CPSI activity. Although the catalysed the NAGS reaction but one of the putative mature existence of the NAGS protein in mammals has been known for enzymes had significantly higher activity than the pre-protein. -

Systems Pathway Engineering of Corynebacterium Crenatum for Improved L-Arginine Production
www.nature.com/scientificreports OPEN Systems pathway engineering of Corynebacterium crenatum for improved L-arginine production Received: 09 March 2016 Zaiwei Man1,*, Meijuan Xu1,*, Zhiming Rao1,2, Jing Guo1, Taowei Yang1, Xian Zhang1 & Accepted: 06 June 2016 Zhenghong Xu3 Published: 24 June 2016 L-arginine is an important amino acid in food and pharmaceutical industries. Until now, the main production method of L-arginine in China is the highly polluting keratin acid hydrolysis. The industrial level L-arginine production by microbial fermentation has become an important task. In previous work, we obtained a new L-arginine producing Corynebacterium crenatum (subspecies of Corynebacterium glutamicum) through screening and mutation breeding. In this work, we performed systems pathway engineering of C. crenatum for improved L-arginine production, involving amplification of L-arginine biosynthetic pathway flux by removal of feedback inhibition and overexpression of arginine operon; optimization of NADPH supply by modulation of metabolic flux distribution between glycolysis and pentose phosphate pathway; increasing glucose consumption by strengthening the preexisting glucose transporter and exploitation of new glucose uptake system; channeling excess carbon flux from glycolysis into tricarboxylic acid cycle to alleviate the glucose overflow metabolism; redistribution of carbon flux atα -ketoglutarate metabolic node to channel more flux into L-arginine biosynthetic pathway; minimization of carbon and cofactor loss by attenuation of byproducts formation. The final strain could produce 87.3 g L−1 L-arginine with yield up to 0.431 g L-arginine g−1 glucose in fed-batch fermentation. L-arginine, a semiessential amino acid, has numerous applications in food flavoring and pharmaceutical indus- tries. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S.