Saccharomyces Cerevisiae Aspartate Kinase Mechanism and Inhibition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METACYC ID Description A0AR23 GO:0004842 (Ubiquitin-Protein Ligase
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Heat Stress Responsive Zostera marina Genes, Southern Population (α=0. -

Hereditary Galactokinase Deficiency J
Arch Dis Child: first published as 10.1136/adc.46.248.465 on 1 August 1971. Downloaded from Alrchives of Disease in Childhood, 1971, 46, 465. Hereditary Galactokinase Deficiency J. G. H. COOK, N. A. DON, and TREVOR P. MANN From the Royal Alexandra Hospital for Sick Children, Brighton, Sussex Cook, J. G. H., Don, N. A., and Mann, T. P. (1971). Archives of Disease in Childhood, 46, 465. Hereditary galactokinase deficiency. A baby with galactokinase deficiency, a recessive inborn error of galactose metabolism, is des- cribed. The case is exceptional in that there was no evidence of gypsy blood in the family concerned. The investigation of neonatal hyperbilirubinaemia led to the discovery of galactosuria. As noted by others, the paucity of presenting features makes early diagnosis difficult, and detection by biochemical screening seems desirable. Cataract formation, of early onset, appears to be the only severe persisting complication and may be due to the biosynthesis and accumulation of galactitol in the lens. Ophthalmic surgeons need to be aware of this enzyme defect, because with early diagnosis and dietary treatment these lens changes should be reversible. Galactokinase catalyses the conversion of galac- and galactose diabetes had been made in this tose to galactose-l-phosphate, the first of three patient (Fanconi, 1933). In adulthood he was steps in the pathway by which galactose is converted found to have glycosuria as well as galactosuria, and copyright. to glucose (Fig.). an unexpectedly high level of urinary galactitol was detected. He was of average intelligence, and his handicaps, apart from poor vision, appeared to be (1) Galactose Gackinase Galactose-I-phosphate due to neurofibromatosis. -

Induction of Uridyl Transferase Mrna-And Dependency on GAL4 Gene Function (In Vitro Translation/Immunoprecipitation/GAL Gene Cluster/Positive Regulation) JAMES E
Proc. Nati. Acad. Sci. USA Vol. 75, No. 6, pp. 2878-2882, June 1978 Genetics Regulation of the galactose pathway in Saccharomyces cerevisiae: Induction of uridyl transferase mRNA-and dependency on GAL4 gene function (in vitro translation/immunoprecipitation/GAL gene cluster/positive regulation) JAMES E. HOPPER*, JAMES R. BROACHt, AND LUCY B. ROWE* * Rosenstiel Basic Medical Sciences Research Center, Brandeis University, Waltham, Massachusetts 02154; and t Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724 Communicated by Norman H. Giles, April 10,1978 ABSTRACT In Saccharomyces cerevisiae, utilization of Genetic control of the inducible galactose pathway enzymes galactose requires four inducible enzyme activities. Three of involves the four structural genes GALI, GAL10, GAL7, and these activities (galactose-l-phosphate uridyl transferase, EC genes, GAL4, GAL81 (c), GAL80 2.7.7.10; uridine diphosphogalactose 4-epimerase, EC 5.1.3.2; GAL2 and four regulatory and galactokinase, EC 2.7.1.6) are specified by three tightly (i), and GALS.* Mutations in GALl, GAL10, GAL7, and GAL2 linked genes (GAL7, GALlO, and GALI, respectively) on chro- affect the individual appearance of galactokinase, epimerase, mosome II, whereas the fourth, galactose transport, is specified transferase, and galactose transport activities, respectively (6). by a gene (GALS) located on chromosome XIL Although classic Mutations defining the GALl, GAL10, and GAL7 genes have genetic analysis has revealed both positive and negative regu- invariably been recessive, and they map in three tightly linked latory genes that coordinately affect the appearance of ail four complementation groups near the centromere of chromosome enzyme activities, neither the basic events leading to the ap- pearance of enzyme activities nor the roles of the regulatory II (6, 9, 10). -

Mapping Active Site Residues in Glutamate-5-Kinase. the Substrate Glutamate and the Feed-Back Inhibitor Proline Bind at Overlapping Sites
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 580 (2006) 6247–6253 Mapping active site residues in glutamate-5-kinase. The substrate glutamate and the feed-back inhibitor proline bind at overlapping sites Isabel Pe´rez-Arellanoa, Vicente Rubiob, Javier Cerveraa,* a Centro de Investigacio´n Prı´ncipe Felipe, Avda. Autopista del Saler, 16, Valencia 46013, Spain b Instituto de Biomedicina de Valencia (IBV-CSIC), Jaime Roig 11, Valencia 46010, Spain Received 22 August 2006; revised 10 October 2006; accepted 12 October 2006 Available online 20 October 2006 Edited by Stuart Ferguson (a domain named after pseudo uridine synthases and archaeo- Abstract Glutamate-5-kinase (G5K) catalyzes the controlling first step of proline biosynthesis. Substrate binding, catalysis sine-specific transglycosylases) domain [10]. AAK domains and feed-back inhibition by proline are functions of the N-termi- bind ATP and a phosphorylatable acidic substrate, and make nal 260-residue domain of G5K. We study here the impact on a phosphoric anhydride [11]. The function of the PUA domain these functions of 14 site-directed mutations affecting 9 residues (a domain characteristically found in some RNA-modifying of Escherichia coli G5K, chosen on the basis of the structure of enzymes) [12] is not known. By deleting the PUA domain of the bisubstrate complex of the homologous enzyme acetylgluta- E. coli G5K we showed [10] that the isolated AAK domain, mate kinase (NAGK). The results support the predicted roles as the complete enzyme, forms tetramers, catalyzes the reac- of K10, K217 and T169 in catalysis and ATP binding and of tion and is feed-back inhibited by proline. -

Adaptive Laboratory Evolution Enhances Methanol Tolerance and Conversion in Engineered Corynebacterium Glutamicum
ARTICLE https://doi.org/10.1038/s42003-020-0954-9 OPEN Adaptive laboratory evolution enhances methanol tolerance and conversion in engineered Corynebacterium glutamicum Yu Wang 1, Liwen Fan1,2, Philibert Tuyishime1, Jiao Liu1, Kun Zhang1,3, Ning Gao1,3, Zhihui Zhang1,3, ✉ ✉ 1234567890():,; Xiaomeng Ni1, Jinhui Feng1, Qianqian Yuan1, Hongwu Ma1, Ping Zheng1,2,3 , Jibin Sun1,3 & Yanhe Ma1 Synthetic methylotrophy has recently been intensively studied to achieve methanol-based biomanufacturing of fuels and chemicals. However, attempts to engineer platform micro- organisms to utilize methanol mainly focus on enzyme and pathway engineering. Herein, we enhanced methanol bioconversion of synthetic methylotrophs by improving cellular tolerance to methanol. A previously engineered methanol-dependent Corynebacterium glutamicum is subjected to adaptive laboratory evolution with elevated methanol content. Unexpectedly, the evolved strain not only tolerates higher concentrations of methanol but also shows improved growth and methanol utilization. Transcriptome analysis suggests increased methanol con- centrations rebalance methylotrophic metabolism by down-regulating glycolysis and up- regulating amino acid biosynthesis, oxidative phosphorylation, ribosome biosynthesis, and parts of TCA cycle. Mutations in the O-acetyl-L-homoserine sulfhydrylase Cgl0653 catalyzing formation of L-methionine analog from methanol and methanol-induced membrane-bound transporter Cgl0833 are proven crucial for methanol tolerance. This study demonstrates the importance of -

| Hai Lui a Un Acutul Luniit Moonhiti
|HAI LUI AUN ACUTULUS010006055B2 LUNIIT MOONHITI (12 ) United States Patent (10 ) Patent No. : US 10 , 006 , 055 B2 Burk et al. (45 ) Date of Patent: Jun . 26 , 2018 ( 54 ) MICROORGANISMS FOR PRODUCING 2002/ 0168654 A1 11/ 2002 Maranas et al. 2003 / 0059792 Al 3 /2003 Palsson et al . BUTADIENE AND METHODS RELATED 2003 /0087381 A1 5 / 2003 Gokarn THERETO 2003 / 0224363 Al 12 /2003 Park et al . 2003 / 0233218 Al 12 /2003 Schilling (71 ) Applicant: Genomatica , Inc. , San Diego , CA (US ) 2004 / 0009466 AL 1 /2004 Maranas et al. 2004 / 0029149 Al 2 /2004 Palsson et al. ( 72 ) Inventors : Mark J . Burk , San Diego , CA (US ) ; 2004 / 0072723 A1 4 /2004 Palsson et al. Anthony P . Burgard , Bellefonte , PA 2004 / 0152159 Al 8 / 2004 Causey et al . 2005 /0042736 A1 2 / 2005 San et al . (US ) ; Robin E . Osterhout , San Diego , 2005 / 0079482 A1 4 / 2005 Maranas et al . CA (US ) ; Jun Sun , San Diego , CA 2006 / 0046288 Al 3 / 2006 Ka - Yiu et al. ( US ) ; Priti Pharkya , San Diego , CA 2006 / 0073577 A1 4 / 2006 Ka - Yiu et al . (US ) 2007 /0184539 Al 8 / 2007 San et al . 2009 / 0047718 Al 2 / 2009 Blaschek et al . 2009 / 0047719 Al 2 / 2009 Burgard et al . (73 ) Assignee : Genomatica , Inc ., San Diego , CA (US ) 2009 /0191593 A1 7 / 2009 Burk et al . 2010 / 0003716 A1 1 / 2010 Cervin et al. ( * ) Notice : Subject to any disclaimer , the term of this 2010 /0184171 Al 7 /2010 Jantama et al. patent is extended or adjusted under 35 2010 /0304453 Al 12 / 2010 Trawick et al . -
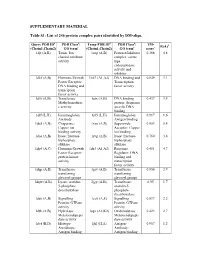
List of 246 Protein Complex Pairs Identified by DM-Align
SUPPLEMENTARY MATERIAL Table S1: List of 246 protein complex pairs identified by DM-align. Query PDB IDa PDB Classb: Temp PDB IDd PDB Classb: TM- R(Å)f (Chain1,Chain2) GO termc (Chain1,Chain2) GO termc scoree 1djt (A,B) Toxin: Ion 1aap (A,B) Protease/Inhibitor 0.368 4.8 channel inhibitor complex: serine activity type endopeptidase activity and inhibitor 1dkf (A,B) Hormone/Growth 1xb7 (A1,A2) DNA binding and 0.849 3.1 Factor Receptor: Transcription DNA binding and factor activity transciption factor activity 1dl5 (A,B) Transferase: 1utx (A,B) DNA binding 0.437 3.9 Methyltransferas protein: Sequence e activity specific DNA binding 1dlf (L,H) Immunoglobin: 1j05 (L,H) Immunoglobin: 0.917 1.6 Antibody Antigen binding 1do5 (A,B) Chaperone: 1xso (A,B) Superoxide 0.953 0.9 Copper ion Acceptor: Copper binding activity ion binding 1dos (A,B) lyase: fructose- 1rvg (A,B) lyase: fructose- 0.760 3.6 biphosphate biphosphate aldolase aldolase 1dp4 (A,C) Hormone/Growth 1dz3 (A1,A2) Response 0.481 4.7 Factor Receptor: Regulator: DNA protein kinase binding and activity transcription factor activity 1dqp (A,B) Transferase: 1grv (A,B) Transferase: 0.856 2.9 transferring transferring glycosyl groups glycosyl groups 1dqw (A,B) Lyase: orotidin- 2jgy (A,B) Transferase: 0.95 1.7 5-phosphate orotidin-5- decarboxylase phosphate decarboxylase 1ds6 (A,B) Signalling 1cc0 (A,E) Signalling 0.897 2.2 Protein: GTPase Protein: GTPase activity activity 1dth (A,B) Hydrolase: 1sqv (A2,K2) Oxidoredutase: 0.423 2.7 Metaloendopepti Metaloendopepti dase activity dase activity 1dvf -

(12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk Et Al
US 2014O155567A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk et al. (43) Pub. Date: Jun. 5, 2014 (54) MICROORGANISMS AND METHODS FOR (60) Provisional application No. 61/331,812, filed on May THE BIOSYNTHESIS OF BUTADENE 5, 2010. (71) Applicant: Genomatica, Inc., San Diego, CA (US) Publication Classification (72) Inventors: Mark J. Burk, San Diego, CA (US); (51) Int. Cl. Anthony P. Burgard, Bellefonte, PA CI2P 5/02 (2006.01) (US); Jun Sun, San Diego, CA (US); CSF 36/06 (2006.01) Robin E. Osterhout, San Diego, CA CD7C II/6 (2006.01) (US); Priti Pharkya, San Diego, CA (52) U.S. Cl. (US) CPC ................. CI2P5/026 (2013.01); C07C II/I6 (2013.01); C08F 136/06 (2013.01) (73) Assignee: Genomatica, Inc., San Diego, CA (US) USPC ... 526/335; 435/252.3:435/167; 435/254.2: (21) Appl. No.: 14/059,131 435/254.11: 435/252.33: 435/254.21:585/16 (22) Filed: Oct. 21, 2013 (57) ABSTRACT O O The invention provides non-naturally occurring microbial Related U.S. Application Data organisms having a butadiene pathway. The invention addi (63) Continuation of application No. 13/101,046, filed on tionally provides methods of using Such organisms to produce May 4, 2011, now Pat. No. 8,580,543. butadiene. Patent Application Publication Jun. 5, 2014 Sheet 1 of 4 US 2014/O155567 A1 ?ueudos!SMS |?un61– Patent Application Publication Jun. 5, 2014 Sheet 2 of 4 US 2014/O155567 A1 VOJ OO O Z?un61– Patent Application Publication US 2014/O155567 A1 {}}} Hººso Patent Application Publication Jun. -
Generate Metabolic Map Poster
Authors: Peter D. Karp Suzanne Paley Julio Collado-Vides John L Ingraham Ingrid Keseler Markus Krummenacker Cesar Bonavides-Martinez Robert Gunsalus An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Carol Fulcher Ian Paulsen Socorro Gama-Castro Robert LaRossa Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. EcoCyc: Escherichia coli K-12 substr. MG1655 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari Amanda Mackie Alberto Santos-Zavaleta succinate phosphate succinate N-acetyl-DL-methionine + L-ornithine glutathione + L-methionine S-oxide D-fructofuranose γ Ag+ molybdate ferroheme b L,L-homocystine asp lys cys L-alanyl- -D- D-mannopyranose 6-phosphate 2+ 2+ H D-methionine 2-deoxy-D-glucose succinate formate formate succinate D-tartrate putrescine agmatine cadaverine L-tartrate D-fructofuranose 6-phosphate + nitrate nitrate Cu thiosulfate deoxycholate L,L-homocystine D-cystine D-cycloserine methyl β-D-glucoside putrescine asp spermidine (S)-2-hydroxybutanoate (S)-2-hydroxybutanoate arg L-homoserine lactone magnesium hydrogenphosphate magnesium hydrogenphosphate antimonous acid glutamyl-meso- Co2+ Cd2+ lactulose poly-β-1,6- met cob(I)inamide 2,3-dioxo- -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Role of Glucokinase and Glucose-6 Phosphatase in the Nutritional Regulation of Endogenous Glucose Production G Mithieux
Role of glucokinase and glucose-6 phosphatase in the nutritional regulation of endogenous glucose production G Mithieux To cite this version: G Mithieux. Role of glucokinase and glucose-6 phosphatase in the nutritional regulation of endogenous glucose production. Reproduction Nutrition Development, EDP Sciences, 1996, 36 (4), pp.357-362. hal-00899845 HAL Id: hal-00899845 https://hal.archives-ouvertes.fr/hal-00899845 Submitted on 1 Jan 1996 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Review Role of glucokinase and glucose-6 phosphatase in the nutritional regulation of endogenous glucose production G Mithieux Unité 197 de l’Inserm, faculté de médecine René-Laënnec, rue Guillaume-Paradin, 69372 Lyon cedex 08, France (Received 29 November 1995; accepted 6 May 1996) Summary ― Two specific enzymes, glucokinase (GK) and glucose-6 phosphatase (Gic6Pase) enable the liver to play a crucial role in glucose homeostasis. The activity of Glc6Pase, which enables the liver to produce glucose, is increased during short-term fasting, in agreement with the enhancement of liver gluconeogenesis. During long-term fasting, Glc6Pase activity is increased in the kidney, which con- tributes significantly to the glucose supply at that time. -
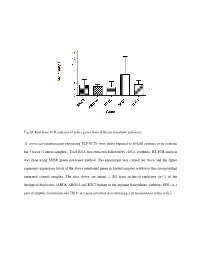
Fig.S1 Real-Time PCR Analysis of Select Genes from Different Metabolic Pathways S. Cerevisiae Transformants Expressing TEF-YCT1
Fig.S1 Real-time PCR analysis of select genes from different metabolic pathways S. cerevisiae transformants expressing TEF-YCT1 were either exposed to 500µM cysteine or no cysteine for 5 hours (Control samples). Total RNA was extracted followed by cDNA synthesis. RT-PCR analysis was done using SYBR green dye-based method. The experiment was carried out twice and the figure represents expression levels of the above mentioned genes in treated samples relative to the corresponding untreated control samples. The data above are means ± SD from technical replicates (n=3) of the biological duplicates. [ARG8, ARG5,6 and RTC2 belong to the arginine biosynthetic pathway, SSU1 is a part of sulphur metabolism and TIS11 is a gene involved in maintaining iron homeostasis in the cells.] Table S1: List of strains used in this study Strain Genotype Source ABC 1738 (yct1) MATα his31 leu20 lys20 ura30 YLL055W:: kanMX4 Euroscarf (Y01543) ABC3612(arg3) MATa his31 leu20 ura30 YJL088w::kanMX4 Euroscarf (Y11335) ABC4812(arg5,6) MATa his31 leu20 ura30 YER069w::kanMX4 Euroscarf (Y10209) ABC4811(arg8) MATa his31 leu20 ura30 YOL140w::kanMX4 Euroscarf (Y17711) ABC4814(spe1) MATa his31 leu20 ura30 YKL184w::kanMX4 Euroscarf (Y15034) ABC4815(spe2) MATa his31 leu20 ura30 YOL052c::kanMX4 Euroscarf (Y11743) ABC4816(spe3) MATa his31 leu20 ura30 YPR069c::kanMX4 Euroscarf (Y15488) ABC4817(spe4) MATa his31 leu20 ura30 YLR146c::kanMX4 Euroscarf (Y16945) ABC3604(ssu1) MATa his31 leu20 ura30 YPL092w::kanMX4 Euroscarf (Y12160) ABC1486(str2) MATa his31 leu20 ura30 YJR130c::kanMX4