Comparative Functional Genomics of Amino Acid Metabolism of Lactic Acid Bacteria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METACYC ID Description A0AR23 GO:0004842 (Ubiquitin-Protein Ligase
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Heat Stress Responsive Zostera marina Genes, Southern Population (α=0. -

The Effect of Selected Herbal Extracts on Lactic Acid Bacteria Activity
applied sciences Article The Effect of Selected Herbal Extracts on Lactic Acid Bacteria Activity Małgorzata Ziarno 1,* , Mariola Kozłowska 2 , Iwona Scibisz´ 3 , Mariusz Kowalczyk 4 , Sylwia Pawelec 4 , Anna Stochmal 4 and Bartłomiej Szleszy ´nski 5 1 Division of Milk Technology, Department of Food Technology and Assessment, Institute of Food Science, Warsaw University of Life Sciences–SGGW (WULS–SGGW), 02-787 Warsaw, Poland 2 Department of Chemistry, Institute of Food Science, Warsaw University of Life Sciences–SGGW (WULS–SGGW), 02-787 Warsaw, Poland; [email protected] 3 Division of Fruit, Vegetable and Cereal Technology, Department of Food Technology and Assessment, Institute of Food Science, Warsaw University of Life Sciences–SGGW (WULS–SGGW), 02-787 Warsaw, Poland; [email protected] 4 Department of Biochemistry and Crop Quality, Institute of Soil Science and Plant Cultivation, State Research Institute, 24-100 Puławy, Poland; [email protected] (M.K.); [email protected] (S.P.); [email protected] (A.S.) 5 Institute of Horticultural Sciences, Warsaw University of Life Sciences–SGGW (WULS–SGGW), 02-787 Warsaw, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-225-937-666 Abstract: This study aimed to investigate the effect of plant extracts (valerian Valeriana officinalis L., sage Salvia officinalis L., chamomile Matricaria chamomilla L., cistus Cistus L., linden blossom Tilia L., ribwort plantain Plantago lanceolata L., marshmallow Althaea L.) on the activity and growth of lactic acid bacteria (LAB) during the fermentation and passage of milk through a digestive system model. Citation: Ziarno, M.; Kozłowska, M.; The tested extracts were also characterized in terms of their content of polyphenolic compounds and Scibisz,´ I.; Kowalczyk, M.; Pawelec, S.; antioxidant activity. -

Kinetics of Ornithine Carbamoyltransferase
15// 454 BIOCHEMICAL SOCIETY TRANSACTIONS Ammonia stimulation of malate dehydrogenase from Bacilllls slearolhermophillls: generation of an apparent aspartate dehydrogenase activity C. N. G. SCHMIDT* and L. JER VISt Attempts to purify aspartate dehydrogenase by salt fraction• • Departmel/t 0/ Botal/Y. Rothamsted Experimel/tal Statiol/. ation. chromatography on Cibacron Blue 3GA-Sepharose or \ Harpel/del/. Herts. A L5 2JQ. U.K .. al/d t Departmel/t 0/ Procion Red HE-3B-Scpharose. affinity chromatography on Biology. Paisley College o/Tee/mology. High Street. Paisley 5'-AMP-Sepharose and gel filtration through Sephacryl S-300 PA I 2BE. Re/l(rewshire. Seollal/d. U.K. always resulted in co-purification of malate dehydrogenase. ' Purification of malate dehydrogenase to near-Ifomogeneity by During investigations into the enzymology of ammonia assimi• the procedure of Wright & Sundaram (1979) yielded a product lation by Baeil/lls stearolhermophillls strain PH24 (Buswell & that was more than 90% pure but still appeared to contain Twomey. 1975). we detected high amounts of an apparent aspartate dehydrogenase in that the addition of ammonia to the aspartate dehydrogenase activity. The enzyme appeared to be malate dehydrogenase assay system caused a 2-fold increase in capable of catalysing the reductive ami nation of oxaloacetic acid the rate of NADH oxidation. Attempts to stain polyacrylamide with NA DH as cofactor. but could not catalyse the oxidative gels for aspartate dehydrogenase activity by using I.-aspartate. deamination of I.-aspartic acid. At low concentrations of NI~4C1 NAD+., Nitro Blue Tetrazolium and phenazonium metho• as the sole nitrogen source in the culture medium. the most sulphate were unsuccessful. -

Mapping Active Site Residues in Glutamate-5-Kinase. the Substrate Glutamate and the Feed-Back Inhibitor Proline Bind at Overlapping Sites
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 580 (2006) 6247–6253 Mapping active site residues in glutamate-5-kinase. The substrate glutamate and the feed-back inhibitor proline bind at overlapping sites Isabel Pe´rez-Arellanoa, Vicente Rubiob, Javier Cerveraa,* a Centro de Investigacio´n Prı´ncipe Felipe, Avda. Autopista del Saler, 16, Valencia 46013, Spain b Instituto de Biomedicina de Valencia (IBV-CSIC), Jaime Roig 11, Valencia 46010, Spain Received 22 August 2006; revised 10 October 2006; accepted 12 October 2006 Available online 20 October 2006 Edited by Stuart Ferguson (a domain named after pseudo uridine synthases and archaeo- Abstract Glutamate-5-kinase (G5K) catalyzes the controlling first step of proline biosynthesis. Substrate binding, catalysis sine-specific transglycosylases) domain [10]. AAK domains and feed-back inhibition by proline are functions of the N-termi- bind ATP and a phosphorylatable acidic substrate, and make nal 260-residue domain of G5K. We study here the impact on a phosphoric anhydride [11]. The function of the PUA domain these functions of 14 site-directed mutations affecting 9 residues (a domain characteristically found in some RNA-modifying of Escherichia coli G5K, chosen on the basis of the structure of enzymes) [12] is not known. By deleting the PUA domain of the bisubstrate complex of the homologous enzyme acetylgluta- E. coli G5K we showed [10] that the isolated AAK domain, mate kinase (NAGK). The results support the predicted roles as the complete enzyme, forms tetramers, catalyzes the reac- of K10, K217 and T169 in catalysis and ATP binding and of tion and is feed-back inhibited by proline. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -
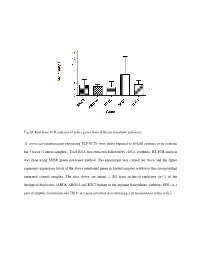
Fig.S1 Real-Time PCR Analysis of Select Genes from Different Metabolic Pathways S. Cerevisiae Transformants Expressing TEF-YCT1
Fig.S1 Real-time PCR analysis of select genes from different metabolic pathways S. cerevisiae transformants expressing TEF-YCT1 were either exposed to 500µM cysteine or no cysteine for 5 hours (Control samples). Total RNA was extracted followed by cDNA synthesis. RT-PCR analysis was done using SYBR green dye-based method. The experiment was carried out twice and the figure represents expression levels of the above mentioned genes in treated samples relative to the corresponding untreated control samples. The data above are means ± SD from technical replicates (n=3) of the biological duplicates. [ARG8, ARG5,6 and RTC2 belong to the arginine biosynthetic pathway, SSU1 is a part of sulphur metabolism and TIS11 is a gene involved in maintaining iron homeostasis in the cells.] Table S1: List of strains used in this study Strain Genotype Source ABC 1738 (yct1) MATα his31 leu20 lys20 ura30 YLL055W:: kanMX4 Euroscarf (Y01543) ABC3612(arg3) MATa his31 leu20 ura30 YJL088w::kanMX4 Euroscarf (Y11335) ABC4812(arg5,6) MATa his31 leu20 ura30 YER069w::kanMX4 Euroscarf (Y10209) ABC4811(arg8) MATa his31 leu20 ura30 YOL140w::kanMX4 Euroscarf (Y17711) ABC4814(spe1) MATa his31 leu20 ura30 YKL184w::kanMX4 Euroscarf (Y15034) ABC4815(spe2) MATa his31 leu20 ura30 YOL052c::kanMX4 Euroscarf (Y11743) ABC4816(spe3) MATa his31 leu20 ura30 YPR069c::kanMX4 Euroscarf (Y15488) ABC4817(spe4) MATa his31 leu20 ura30 YLR146c::kanMX4 Euroscarf (Y16945) ABC3604(ssu1) MATa his31 leu20 ura30 YPL092w::kanMX4 Euroscarf (Y12160) ABC1486(str2) MATa his31 leu20 ura30 YJR130c::kanMX4 -

Antifungal Activity of Lactobacillus Pentosus ŁOCK 0979 in the Presence of Polyols and Galactosyl-Polyols
Probiotics & Antimicro. Prot. https://doi.org/10.1007/s12602-017-9344-0 Antifungal Activity of Lactobacillus pentosus ŁOCK 0979 in the Presence of Polyols and Galactosyl-Polyols Lidia Lipińska1 & Robert Klewicki2 & Michał Sójka2 & Radosław Bonikowski3 & Dorota Żyżelewicz2 & Krzysztof Kołodziejczyk2 & Elżbieta Klewicka 1 # The Author(s) 2017. This article is an open access publication Abstract The antifungal activity of Lactobacillus pentosus Keywords Antifungal activity . Galactosyl-polyols . ŁOCK 0979 depends both on the culture medium and on the Lactobacillus . Metabolites . Polyols . SEM fungal species. In the control medium, the strain exhibited limited antagonistic activity against indicator food-borne molds and yeasts. However, the supplementation of the bac- Introduction terial culture medium with polyols (erythritol, lactitol, maltitol, mannitol, sorbitol, xylitol) or their galactosyl deriva- Filamentous fungi and yeasts are present in almost all types of tives (gal-erythritol, gal-sorbitol, gal-xylitol) enhanced the an- ecosystems due to their high adaptation ability and low nutri- tifungal properties of Lactobacillus pentosus ŁOCK 0979. Its tional requirements. Filamentous fungi are widespread food metabolites were identified and quantified by enzymatic spoilage microorganisms responsible for significant economic methods, HPLC, UHPLC-MS coupled with QuEChERS, losses in the agri-food industry [6]; they are also a major and GC-MS. The presence of polyols and gal-polyols signif- health concern due to mycotoxin production. The most com- icantly affected the acid metabolite profile of the bacterial mon genera of spoilage fungi include Penicillium, Fusarium, culture supernatant. In addition, lactitol and mannitol were Aspergillus, Cladosporium,andRhizopus [21]. Commercial used by bacteria as alternative carbon sources. A number of foodstuffs are usually protected from such microorganisms by compounds with potential antifungal properties were identi- physical and chemical techniques. -

The Impact of Oil Type and Lactic Acid Bacteria on Conjugated Linoleic Acid Production
JOBIMB, 2016, Vol 4, No 2, 25-29 JOURNAL OF BIOCHEMISTRY, MICROBIOLOGY AND BIOTECHNOLOGY Website: http://journal.hibiscuspublisher.com/index.php/JOBIMB/index The Impact of Oil Type and Lactic Acid Bacteria on Conjugated Linoleic Acid Production Mahmoud A. Al-Saman 1*, Rafaat M. Elsanhoty 1 and Elhadary A. E. 2 1Department of Industrial Biotechnology, Genetic Engineering and Biotechnology Research Institute, University of Sadat City, Sadat City 22857/79, Egypt. 2Biochemistry Department, Faculty of Agriculture, Benha University, Egypt. *Corresponding author: Dr. Mahmoud Abd El-Hamid Al-Saman Department of Industrial Biotechnology, Genetic Engineering and Biotechnology Research Institute, University of Sadat City, Sadat City 22857/79, Egypt. Email: [email protected] [email protected] HISTORY ABSTRACT This work was conducted to investigate the effect of oil type and lactic acid bacteria on the Received: 27 th October 2016 conjugated linoleic acid (CLA) production in MRS medium. The ability of eight strains of Received in revised form: 2nd December 2016 Accepted: 17th December 2016 lactic acids bacteria; Lactobacillus acidophilus (P2, ATCC 20552), Lactobacillus brevis (P102), Lactobacillus casei (P9, DSMZ 20011), Lactobacillus plantarum (P1), Lactobacillus KEYWORDS pentosus (P4), Lactobacillus rhamnosus (P5, TISTR 541), Bifidobacterium longum (BL) and conjugated linoleic acid (CLA) Bifidobacterium lactis (P7, Bb-12) for the production of CLA in the MRS broth was lactic acid bacteria investigated. Two vegetable oils (sun flower oil & linseed oil) and cod liver oil were used as vegetable oils cod liver oil substrates in MRS media. The oils were added to MRS in concentration of 10 mg/ml and probiotic incubated for three days at 37°C. -

Effect of Short Time Exposure of Rats to Extreme Low Temperature on Some Plasma and Liver Enzymes
Bull Vet Inst Pulawy 50, 121-124, 2006 EFFECT OF SHORT TIME EXPOSURE OF RATS TO EXTREME LOW TEMPERATURE ON SOME PLASMA AND LIVER ENZYMES EWA ROMUK1, EWA BIRKNER1, BRONISŁAWA SKRZEP-POLOCZEK1, LESZEK JAGODZIŃSKI2, AGATA STANEK2, BERNADETA WIŚNIOWSKA3 AND ALEKSANDER SIEROŃ2 1Department of Biochemistry in Zabrze, 2Clinic of Internal Diseases, Angiology and Physical Medicine in Bytom, Medical University of Silesia, 40-006 Katowice, Poland 3Center of Cryotherapy, 41-709 Ruda Śląska, Poland e-mail: [email protected] Received for publication October 10, 2005. Abstract protocol of the studies was reviewed and approved by the Bioethical Committee of Medical University of The aim of the study was to search the influence of Silesia in Katowice. Animals were supplied by the cryotherapy on liver enzyme activity in experimental rat Experimental Animal Farm. The rats underwent two model. The first group of rats was exposed 1 min daily to - week environmental adaptation cycle. After this period 90°C for 5 d, the second group was exposed 1 min daily to - the animals were randomized into 3 equal groups. Group 90°C for 10 d and the control group was not exposed to low I - rats exposed to -90°C for 5 d; group II - rats exposed temperature. A statistically significant increase in the activity to -90°C for 10 d and group III - control rats – without of glutamate dehydrogenase, sorbitol dehydrogenase, malate exposure to cryotherapy. All the groups were fed dehydrogenase, ornithine transcarbamoylase and arginase was observed in the plasma and liver. The obtained results indicate standard rat chow ad libitum. the influence of low temperature on liver metabolism. -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

A Physiological Comparative Study of Acid Tolerance of Lactobacillus Plantarum ZDY 2013 and L
Ann Microbiol (2017) 67:669–677 DOI 10.1007/s13213-017-1295-x ORIGINAL ARTICLE A physiological comparative study of acid tolerance of Lactobacillus plantarum ZDY 2013 and L. plantarum ATCC 8014 at membrane and cytoplasm levels Yilin Guo 1 & Ximei Tian1 & Renhui Huang1 & Xueying Tao1 & Nagendra P. Shah2 & Hua Wei1,3 & Cuixiang Wan3 Received: 8 April 2017 /Accepted: 4 August 2017 /Published online: 23 August 2017 # Springer-Verlag GmbH Germany and the University of Milan 2017 Abstract This study aimed to disclose the acid tolerance metabolism, increased amino acid and enzyme level) of mechanism of Lactobacillus plantarum by comparing L. plantarum ZDY 2013 can protect the cells from acid stress. L. plantarum ZDY 2013 with the type strain L. plantarum ATCC 8014 in terms of cell membrane, energy metabolism, Keywords Lactobacillus plantarum . Acid tolerance . Cell and amino acid metabolism. L. plantarum ZDY 2013 had a membrane . Energy metabolism . Amino acids superior growth performance under acidic condition with 100-fold higher survival rate than that of L. plantarum ATCC 8014 at pH 2.5. To determine the acid tolerance Introduction physiological mechanism, cell integrity was investigated through scanning electron microscopy. The study revealed Lactic acid bacteria (LAB) have been used to produce that L. plantarum ZDY 2013 maintained cell morphology fermented food over the past decades and have been de- and integrity, which is much better than L. plantarum veloped as probiotics, which are generally recognized as ATCC 8014 under acid stress. Analysis of energy metabo- safe (GRAS) (De Vries et al. 2006), for their health- lism showed that, at pH 5.0, L. -

Methods of Extraction, Refining and Concentration of Fish Oil As a Source of Omega-3 Fatty Acids
Corpoica Cienc Tecnol Agropecuaria, Mosquera (Colombia), 19(3):645-668 september - december / 2018 ISSN 0122-8706 ISSNe 2500-5308 645 Transformation and agro-industry Review article Methods of extraction, refining and concentration of fish oil as a source of omega-3 fatty acids Métodos de extracción, refinación y concentración de aceite de pescado como fuente de ácidos grasos omega 3 Jeimmy Rocío Bonilla-Méndez,1* José Luis Hoyos-Concha2 1 Researcher, Universidad del Cauca, Facultad de Ciencias Agrarias. Popayán, Colombia. Email: [email protected]. orcid.org/0000-0001-5362-5950 2 Lecturer, Universidad del Cauca, Facultad de Ciencias Agrarias. Popayán, Colombia. Email: [email protected]. orcid.org/0000-0001-9025-9734 Editor temático: Miguel Ángel Rincón Cervera (Instituto de Nutrición y Tecnología de los Alimentos [INTA]) Date of receipt: 05/07/2017 Date of approval: 15/03/2018 How to cite this article: Bonilla-Méndez, J. R., & Hoyos-Concha, J. L. (2018). Methods of extraction, refining and concentration of fish oil as a source of omega-3 fatty acids. Corpoica Ciencia y Tecnología Agropecuaria, 19(3), 645-668. DOI: https://doi.org/10.21930/rcta.vol19_num2_art:684 This license allows distributing, remixing, retouching, and creating from the work in a non-commercial manner, as long as credit is given and their new creations are licensed under the same conditions. * Corresponding author. Universidad del Cauca, Facultad de Ciencias Agrarias. Vereda Las Guacas, Popayán, Colombia. 2018 Corporación Colombiana de Investigación Agropecuaria Corpoica Cienc Tecnol Agropecuaria, Mosquera (Colombia), 19(3):645-668 september - december / 2018 ISSN 0122-8706 ISSNe 2500-5308 Abstract Fish oil is an industrial product of high nutritional methods, there are new technologies with potential value because of its Omega-3 polyunsaturated fatty to be applied on fish oil.