Andersen-Tawil Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non Commercial Use Only
Cardiogenetics 2017; volume 7:6304 Sudden death in a young patient with atrial fibrillation Case Report Correspondence: María Angeles Espinosa Castro, Inherited Cardiovascular Disease A 22-year-old man suffered a sudden Program, Cardiology Department, Gregorio María Tamargo, cardiac arrest without previous symptoms Marañón Hospital, Dr. Esquerdo, 46, 28007, María Ángeles Espinosa, while he was at rest, waiting for a subway Madrid, Spain. Víctor Gómez-Carrillo, Miriam Juárez, train. Cardiopulmonary resuscitation was Tel.: +34.91.586.82.90. immediately started using an Automated E-mail: [email protected] Francisco Fernández-Avilés, External Defibrillation that identified the Raquel Yotti Key words: KCNQ1; mutation; channelopa- presence of ventricular fibrillation and thy; sudden cardiac death; atrial fibrillation. Inherited Cardiovascular Disease delivered a shock. Return of spontaneous Program, Cardiology Department, circulation was achieved after three Contributions: MT, acquisition and interpreta- Gregorio Marañón Hospital, Madrid, attempts, being atrial fibrillation (AF) the tion of data for the work, ensuring that ques- Spain patient’s rhythm at this point (Figure 1). tions related to the accuracy or integrity of any He was admitted to our Cardiovascular part of the work is appropriately investigated Intensive Care Unit and therapeutic and resolved; MAE, conception of the work, hypothermia was performed over a period critical revision of the intellectual content, final approval of the version to be published, Abstract of 24 h. After completing hypothermia, ensuring that questions related to the accuracy rewarming, and another 24 h of controlled of any part of the work is appropriately inves- Sudden cardiac death (SCD) in young normothermia the patient awakened with no tigated and resolved; VG-C, acquisition and patients without structural heart disease is residual neurologic damage. -
Long QT Syndrome Testing
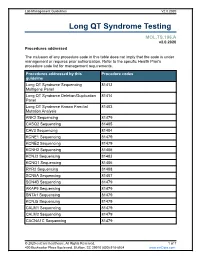
Lab Management Guidelines V2.0.2020 Long QT Syndrome Testing MOL.TS.196.A v2.0.2020 Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline Long QT Syndrome Sequencing 81413 Multigene Panel Long QT Syndrome Deletion/Duplication 81414 Panel Long QT Syndrome Known Familial 81403 Mutation Analysis ANK2 Sequencing 81479 CASQ2 Sequencing 81405 CAV3 Sequencing 81404 KCNE1 Sequencing 81479 KCNE2 Sequencing 81479 KCNH2 Sequencing 81406 KCNJ2 Sequencing 81403 KCNQ1 Sequencing 81406 RYR2 Sequencing 81408 SCN5A Sequencing 81407 SCN4B Sequencing 81479 AKAP9 Sequencing 81479 SNTA1 Sequencing 81479 KCNJ5 Sequencing 81479 CALM1 Sequencing 81479 CALM2 Sequencing 81479 CACNA1C Sequencing 81479 © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V2.0.2020 What is Long QT syndrome Definition Long QT Syndrome (LQTS) is caused by mutations in a number of genes, most of which are related to the functioning of sodium or potassium ion channels in the heart.1 Testing may offer prognostic information in some cases, as specific genes and even specific mutations within those genes may have some correlation to risk for sudden death, effectiveness of beta-blocker therapy, and preventive strategies.1-4 Signs and symptoms of long QT syndrome (LQTS) are variable, but may include a prolonged QT interval on an electrocardiogram, torsades de pointes, syncope, seizures, cardiac arrest, and sudden cardiac death.1,2 Many patients with LQTS can be largely asymptomatic, with cardiac arrest or sudden cardiac death as the first and only symptom. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627). -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?
The Science Journal of the Lander College of Arts and Sciences Volume 11 Number 1 Fall 2017 - 2017 Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Touro College Follow this and additional works at: https://touroscholar.touro.edu/sjlcas Part of the Cardiovascular Diseases Commons, and the Medical Genetics Commons Recommended Citation Braun, M. (2017). Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?. The Science Journal of the Lander College of Arts and Sciences, 11(1). Retrieved from https://touroscholar.touro.edu/sjlcas/vol11/iss1/8 This Article is brought to you for free and open access by the Lander College of Arts and Sciences at Touro Scholar. It has been accepted for inclusion in The Science Journal of the Lander College of Arts and Sciences by an authorized editor of Touro Scholar. For more information, please contact [email protected]. Should Genetic Testing be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Menachem Braun graduated with a BS in Biology in September 2017. Abstract 7KH/RQJ476\QGURPH /476 LVDIDPLOLDOSRWHQWLDOO\IDWDOFDUGLDFDUUK\WKPLD7UDGLWLRQDOO\LWKDVEHHQGLDJQRVHGE\(&* Molecular studies have provided evidence that LQTS can be caused by a range of underlying molecular abnormalities. Genetic research has proven that different forms of LQTS have different genotypic bases. Therefore, it has become possible to diagnose WKHVSHFLÀFW\SHRIGLVHDVHJHQHWLFDOO\7KLVVWXG\H[DPLQHVWKHDGYDQFHPHQWVPDGHLQWKHSDVWWKLUW\\HDUVLQXQGHUVWDQGLQJ /476DQGUHVHDUFKUHJDUGLQJWKHXVHRIJHQHWLFWHVWLQJLQRUGHUWRGHWHUPLQHWKHEHQHÀWVRIJHQHWLFWHVWLQJIRUWKLVGLVHDVH$ survey of original studies which produced the information is presented here, and provides the reader with an understanding of the PHFKDQLFVRIWKHGLVHDVHDQGKRZWKH\GLIIHULQWKHVHYHUDOJHQHWLFYDULDQWV5HVHDUFKVKRZVWKDWWKHEHQHÀWRIJHQHWLFWHVWLQJ must be weighed against the personal implications in may have for a particular patient and his or her family. -

Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes
Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes Michael J. Ackerman, MD, PhD Windland Smith Rice Cardiovascular Genomics Research Professor Professor of Medicine, Pediatrics, and Pharmacology Director, Long QT Syndrome Clinic and the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory President, Sudden Arrhythmia Death Syndromes (SADS) Foundation Learning Objectives to Disclose: • To RECOGNIZE the “faces” (phenotypes) of the congenital long QT syndromes (LQTS) • To CRITIQUE the various diagnostic modalities used in the evaluation of LQTS and UNDERSTAND their limitations • To ASSESS the currently available treatment options for the various LQT syndromes and EVALUATE their efficacy WINDLAND Smith Rice Sudden Death Genomics Laboratory Conflicts of Interest to Disclose: • Consultant – Boston Scientific, Gilead Sciences, Medtronic, St. Jude Medical, and Transgenomic/FAMILION • Royalties – Transgenomic/FAMILION Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT ♥ 1957 – first clinical description – JLNS ♥ 1960s – RomanoProlonged-Ward QT syndrome ♥ 1983 – “Schwartz/Moss score”1. Syncope ♥ 1991 – first LQTS chromosome locus 2. Seizures ♥ March 10, 1995 – birth of cardiac 3. Sudden channelopathies death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal -

Long QT Syndrome
orphananesthesia Anaesthesia recommendations for patients suffering from Long QT syndrome Disease name: Long QT syndrome ICD 10: I45.8 Synonyms: LQTS Related syndromes: Romano-Ward S., Jervell and Lange-Nielsen S., Timothy S. Long QT syndrome (LQTS) is a cardiac disorder resulting from malfunction of cardiac ion channels. It can be divided in congenital (c-LQTS) and acquired (a-LQTS) forms. Typical is a prolongation of the QT interval on ECG and a propensity to ventricular tachyarrhythmias which can lead to a characteristic polymorphic ventricular tachycardia known as “torsades de pointes”. There is a risk of syncope, cardiac arrest or sudden death. The diagnosis is based on ECG findings, clinical and family history. The Schwartz score defines the probability of LQTS according to three categories: 1 point, low probability of LQTS; (2) 2 or 3 points, intermediate probability of LQTS; and (3) ≥3.5 points, high probability of LQTS. THE TABLE OF THE SCHWARTZ SCORE should be added here: Table 2 of the paper Schwartz PJ and Crotti L. QTc behavior during exercise and genetic testing for the Long-QT Syndrome: Circulation 2011;124:2181-2184. Medicine in progress Perhaps new knowledge Every patient is unique Perhaps the diagnostic is wrong Find more information on the disease, its centres of reference and patient organisations on Orphanet: www.orpha.net 1 Typical surgery The typical surgery for LQTS is left cardiac sympathetic denervation, which is recommended in patients symptomatic for cardiac events despite beta-blocker (BB) therapy or in patients in whom BBs are contraindicated or not tolerated. For therapeutic reasons the implantation of a cardioverter defibrillator (ICD) is recommended in patients with a previous cardiac arrest or in patients with recurrent syncopal events despite antiadrenergic interventions. -

Update on the Diagnosis and Management of Familial Long QT Syndrome
Heart, Lung and Circulation (2016) 25, 769–776 POSITION STATEMENT 1443-9506/04/$36.00 http://dx.doi.org/10.1016/j.hlc.2016.01.020 Update on the Diagnosis and Management of Familial Long QT Syndrome Kathryn E Waddell-Smith, FRACP a,b, Jonathan R Skinner, FRACP, FCSANZ, FHRS, MD a,b*, members of the CSANZ Genetics Council Writing Group aGreen Lane Paediatric and Congenital Cardiac Services, Starship Children’s Hospital, Auckland New Zealand bDepartment[5_TD$IF] of Paediatrics,[6_TD$IF] Child[7_TD$IF] and[8_TD$IF] Youth[9_TD$IF] Health,[10_TD$IF] University of Auckland, Auckland, New Zealand Received 17 December 2015; accepted 20 January 2016; online published-ahead-of-print 5 March 2016 This update was reviewed by the CSANZ Continuing Education and Recertification Committee and ratified by the CSANZ board in August 2015. Since the CSANZ 2011 guidelines, adjunctive clinical tests have proven useful in the diagnosis of LQTS and are discussed in this update. Understanding of the diagnostic and risk stratifying role of LQTS genetics is also discussed. At least 14 LQTS genes are now thought to be responsible for the disease. High-risk individuals may have multiple mutations, large gene rearrangements, C-loop mutations in KCNQ1, transmembrane mutations in KCNH2, or have certain gene modifiers present, particularly NOS1AP polymorphisms. In regards to treatment, nadolol is preferred, particularly for long QT type 2, and short acting metoprolol should not be used. Thoracoscopic left cardiac sympathectomy is valuable in those who cannot adhere to beta blocker therapy, particularly in long QT type 1. Indications for ICD therapies have been refined; and a primary indication for ICD in post-pubertal females with long QT type 2 and a very long QT interval is emerging. -

Romano-Ward Syndrome
Romano-Ward syndrome Description Romano-Ward syndrome is a condition that causes a disruption of the heart's normal rhythm (arrhythmia). This disorder is a form of long QT syndrome, which is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. The term "long QT" refers to a specific pattern of heart activity that is detected with an electrocardiogram (ECG or EKG), which is a test used to measure the electrical activity of the heart. In people with long QT syndrome, the part of the heartbeat known as the QT interval is abnormally long. Abnormalities in the time it takes to recharge the heart lead to abnormal heart rhythms. The arrhythmia associated with Romano-Ward syndrome can lead to fainting (syncope) or cardiac arrest and sudden death. However, some people with Romano-Ward syndrome never experience any health problems associated with the condition. Fifteen types of long QT syndrome have been defined based on their genetic cause. Some types of long QT syndrome involve other cardiac abnormalities or problems with additional body systems. Romano-Ward syndrome encompasses those types that involve only a long QT interval without other abnormalities. Frequency Romano-Ward syndrome is the most common form of inherited long QT syndrome, which affects an estimated 1 in 2,000 people worldwide. Long QT syndrome may actually be more common than this estimate, however, because some people never experience any symptoms associated with arrhythmia and therefore may not be diagnosed. Causes Mutations in the KCNQ1, KCNH2, and SCN5A genes are the most common causes of Romano-Ward syndrome. -

Diagnosis, Management and Therapeutic Strategies for Congenital
Review Diagnosis, management and therapeutic Heart: first published as 10.1136/heartjnl-2020-318259 on 26 May 2021. Downloaded from strategies for congenital long QT syndrome Arthur A M Wilde , Ahmad S Amin, Pieter G Postema Heart Centre, Department ABSTRACT Diagnosis of Cardiology, Amsterdam Congenital long QT syndrome (LQTS) is characterised The diagnosis of LQTS relies on the heart rate Universitair Medische Centra, Amsterdam, The Netherlands by heart rate corrected QT interval prolongation and corrected QT interval (QTc) and on a number of life- threatening arrhythmias, leading to syncope and other electrocardiographic parameters as well as Correspondence to sudden death. Variations in genes encoding for cardiac elements obtained by history taking (eg, symptoms Professor Arthur A M Wilde, ion channels, accessory ion channel subunits or proteins and family history). Together they form the LQTS Amsterdam Universitair modulating the function of the ion channel have been probability or Schwartz score, where a score of Medische Centra, Amsterdam, identified as disease-causing mutations in up to 75% of ≥3.5 points indicates a high probability of LQTS The Netherlands; 2 3 a. a. wilde@ amsterdamumc. nl all LQTS cases. Based on the underlying genetic defect, (figures 1 and 2). Genetic information is not LQTS has been subdivided into different subtypes. part of the Schwartz score but an individual with Received 25 January 2021 Growing insights into the genetic background and a pathogenic variant also fulfills the current diag- Revised 12 April -

NEW Long QT Syndrome Booklet.Indd
Long QT Syndrome The Heart Rhythm Charity Promoting better understanding, diagnosis, treatment and quality of life for individuals with cardiac arrhythmias Long QT Syndrome – Patient Information Leaflet www.heartrhythmcharity.org.uk Registered Charity No. 1107496 ©2008 Introduction to Long QT Syndrome This booklet is intended for use by people who wish to understand more about Long QT Syndrome. The information within this booklet comes from research and previous patients experiences. This booklet offers an explanation of the Long QT condition. Additional information can be sourced from the website www.heartrhythmcharity.org.uk Arrhythmia Alliance (A-A) is a coalition of charities, patient groups, patients, carers, medical groups and allied professionals. These groups remain independent, however, work together under the A-A umbrella to promote timely and effective diagnosis and treatment of arrhythmias. A-A supports and promotes the aims and objectives of the individual groups. Contents Glossary Introduction Arrhythmia Heart rhythm disorder What is Long QT Syndrome? What is the QT interval? Cardiologist A doctor who has specialised in What are the symptoms? the diagnosis and treatment of patients with a heart condition What causes Long QT Syndrome Electrocardiogram (ECG) A 12 lead recording of the What are the risks? activity of the heart Test and Diagnosis Implantable Cardioverter Medical History and Assessment Defibrillator (ICD) A small device containing a Genetic Testing battery connected to your heart for monitoring your heart rhythm Treatment Lifestyle Adjustments, Pacemaker after treatment A small metal device implanted under the skin, which produces War ning electrical impulses to treat an abnormal heart rhythm Useful websites Further reading Syncope A blackout/faint due to a sudden lack of blood supply to the brain Arrhythmia Alliance patient booklets are reviewed annually. -

HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes
This document is embargoed until May 10, 2013 at 9:30 am MST. Document will publish in HeartRhythm, EP Europace, and Journal of Arrhythmias in the fall of 2013. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes Developed in partnership with the Heart Rhythm Society (HRS), the European Heart Rhythm Association (EHRA), a registered branch of the European Society of Cardiology, and the Asia Pacific Heart Rhythm Society (APHRS); and in collaboration with the American College of Cardiology Foundation (ACCF), the American Heart Association (AHA), the Pediatric and Congenital Electrophysiology Society (PACES) and the Association for European Pediatric and Congenital Cardiology (AEPC). Document endorsed by HRS, EHRA and APHRS May, 2013. Endorsements pending from ACCF, AHA, PACES, and AEPC. Writing Group Chairs Silvia G. Priori, MD, PhD, Fondazione Salvatore Maugeri, Department of Molecular Medicine University of Pavia, Pavia, ITALY (HRS Chairperson) Arthur A. Wilde, MD, PhD, University of Amsterdam, Amsterdam, NETHERLANDS (EHRA Chairperson) Minoru Horie, MD, PhD, Shiga University of Medical Sciences, Otsu, JAPAN (APHRS Chairperson) Yongkeun Cho, MD, PhD, Kyungpook National University, Daegu, SOUTH KOREA (APHRS Chairperson) Writing Group Members Elijah R. Behr, MA, MBBS, MD, FRCP, St. Georges University of London, UNITED KINGDOM Charles Berul, MD, FHRS, CCDS, Children’s National Medical Center, Washington, DC, UNITED STATES Nico Blom, MD, PhD, Academical Medical Center, AMSTERDAM, Leiden University Medical Center, Leiden, NETHERLANDS* Josep Brugada, MD, PhD, University of Barcelona, Barcelona, SPAIN Chern-En Chiang, MD, PhD, Taipei Veteran’s General Hospital, Taipei, TAIWAN © Heart Rhythm Society, European Heart Rhythm Association, a registered branch of the European Society of Cardiology, and the Asia Pacific Heart Rhythm Society 1 This document is embargoed until May 10, 2013 at 9:30 am MST.