HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non Commercial Use Only
Cardiogenetics 2017; volume 7:6304 Sudden death in a young patient with atrial fibrillation Case Report Correspondence: María Angeles Espinosa Castro, Inherited Cardiovascular Disease A 22-year-old man suffered a sudden Program, Cardiology Department, Gregorio María Tamargo, cardiac arrest without previous symptoms Marañón Hospital, Dr. Esquerdo, 46, 28007, María Ángeles Espinosa, while he was at rest, waiting for a subway Madrid, Spain. Víctor Gómez-Carrillo, Miriam Juárez, train. Cardiopulmonary resuscitation was Tel.: +34.91.586.82.90. immediately started using an Automated E-mail: [email protected] Francisco Fernández-Avilés, External Defibrillation that identified the Raquel Yotti Key words: KCNQ1; mutation; channelopa- presence of ventricular fibrillation and thy; sudden cardiac death; atrial fibrillation. Inherited Cardiovascular Disease delivered a shock. Return of spontaneous Program, Cardiology Department, circulation was achieved after three Contributions: MT, acquisition and interpreta- Gregorio Marañón Hospital, Madrid, attempts, being atrial fibrillation (AF) the tion of data for the work, ensuring that ques- Spain patient’s rhythm at this point (Figure 1). tions related to the accuracy or integrity of any He was admitted to our Cardiovascular part of the work is appropriately investigated Intensive Care Unit and therapeutic and resolved; MAE, conception of the work, hypothermia was performed over a period critical revision of the intellectual content, final approval of the version to be published, Abstract of 24 h. After completing hypothermia, ensuring that questions related to the accuracy rewarming, and another 24 h of controlled of any part of the work is appropriately inves- Sudden cardiac death (SCD) in young normothermia the patient awakened with no tigated and resolved; VG-C, acquisition and patients without structural heart disease is residual neurologic damage. -

Characterization of Atrial Fibrillation Adverse Events Reported in Ibrutinib Randomized Controlled Registration Trials
Complications in Hematology SUPPLEMENTARY APPENDIX Characterization of atrial fibrillation adverse events reported in ibrutinib randomized controlled registration trials Jennifer R. Brown, 1 Javid Moslehi, 2 Susan O’Brien, 3 Paolo Ghia, 4 Peter Hillmen, 5 Florence Cymbalista, 6 Tait D. Shanafelt, 7 Graeme Fraser, 8 Simon Rule, 9 Thomas J. Kipps, 10 Steven Coutre, 11 Marie-Sarah Dilhuydy, 12 Paula Cramer, 13 Alessandra Tedeschi, 14 Ulrich Jaeger, 15 Martin Dreyling, 16 John C. Byrd, 17 Angela Howes, 18 Michael Todd, 19 Jessica Ver - meulen, 20 Danelle F. James, 21 Fong Clow, 21 Lori Styles, 21 Rudy Valentino, 21 Mark Wildgust, 19 Michelle Mahler 19 and Jan A. Burger 22 1Dana-Farber Cancer Institute, Boston, MA, USA; 2Division of Cardiovascular Medicine and Cardio-Oncology Program Vanderbilt School of Medicine, Nashville, TN, USA; 3Chao Family Comprehensive Cancer Center, University of California, Irvine, Orange, CA, USA; 4Univer - sità Vita-Salute San Raffaele and IRCCS Istituto Scientifico San Raffaele, Milano, Italy; 5CA Leeds Teaching Hospitals, St. James Insti - tute of Oncology, Leeds, UK; 6Hôpital Avicenne, AP-HP, UMR Paris13/INSERM U978, Bobigny, France; 7Mayo Clinic, Rochester, MN, USA; 8Juravinski Cancer Centre, McMaster University, Hamilton, ON, Canada; 9Department of Haematology, Plymouth University Med - ical School, Plymouth, UK; 10 Moores UCSD Cancer Center, San Diego, CA, USA; 11 Stanford University School of Medicine and Stanford Cancer Institute, Stanford, CA, USA; 12 Hôpital Haut-Lévêque, Bordeaux, Pessac, France; 13 -

Antithrombotic Therapy in Atrial Fibrillation Associated with Valvular Heart Disease
Europace (2017) 0, 1–21 EHRA CONSENSUS DOCUMENT doi:10.1093/europace/eux240 Antithrombotic therapy in atrial fibrillation associated with valvular heart disease: a joint consensus document from the European Heart Rhythm Association (EHRA) and European Society of Cardiology Working Group on Thrombosis, endorsed by the ESC Working Group on Valvular Heart Disease, Cardiac Arrhythmia Society of Southern Africa (CASSA), Heart Rhythm Society (HRS), Asia Pacific Heart Rhythm Society (APHRS), South African Heart (SA Heart) Association and Sociedad Latinoamericana de Estimulacion Cardıaca y Electrofisiologıa (SOLEACE) Gregory Y. H. Lip1*, Jean Philippe Collet2, Raffaele de Caterina3, Laurent Fauchier4, Deirdre A. Lane5, Torben B. Larsen6, Francisco Marin7, Joao Morais8, Calambur Narasimhan9, Brian Olshansky10, Luc Pierard11, Tatjana Potpara12, Nizal Sarrafzadegan13, Karen Sliwa14, Gonzalo Varela15, Gemma Vilahur16, Thomas Weiss17, Giuseppe Boriani18 and Bianca Rocca19 Document Reviewers: Bulent Gorenek20 (Reviewer Coordinator), Irina Savelieva21, Christian Sticherling22, Gulmira Kudaiberdieva23, Tze-Fan Chao24, Francesco Violi25, Mohan Nair26, Leandro Zimerman27, Jonathan Piccini28, Robert Storey29, Sigrun Halvorsen30, Diana Gorog31, Andrea Rubboli32, Ashley Chin33 and Robert Scott-Millar34 * Corresponding author. Tel/fax: þ44 121 5075503. E-mail address: [email protected] Published on behalf of the European Society of Cardiology. All rights reserved. VC The Author 2017. For permissions, please email: [email protected]. 2 G.Y.H. Lip 1Institute of Cardiovascular Sciences, University of Birmingham and Aalborg Thrombosis Research Unit, Department of Clinical Medicine, Aalborg University, Denmark (Chair, representing EHRA); 2Sorbonne Universite´ Paris 6, ACTION Study Group, Institut De Cardiologie, Groupe Hoˆpital Pitie´-Salpetrie`re (APHP), INSERM UMRS 1166, Paris, France; 3Institute of Cardiology, ‘G. -
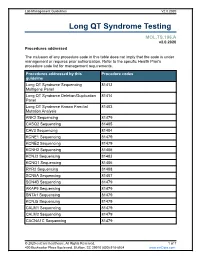
Long QT Syndrome Testing
Lab Management Guidelines V2.0.2020 Long QT Syndrome Testing MOL.TS.196.A v2.0.2020 Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline Long QT Syndrome Sequencing 81413 Multigene Panel Long QT Syndrome Deletion/Duplication 81414 Panel Long QT Syndrome Known Familial 81403 Mutation Analysis ANK2 Sequencing 81479 CASQ2 Sequencing 81405 CAV3 Sequencing 81404 KCNE1 Sequencing 81479 KCNE2 Sequencing 81479 KCNH2 Sequencing 81406 KCNJ2 Sequencing 81403 KCNQ1 Sequencing 81406 RYR2 Sequencing 81408 SCN5A Sequencing 81407 SCN4B Sequencing 81479 AKAP9 Sequencing 81479 SNTA1 Sequencing 81479 KCNJ5 Sequencing 81479 CALM1 Sequencing 81479 CALM2 Sequencing 81479 CACNA1C Sequencing 81479 © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V2.0.2020 What is Long QT syndrome Definition Long QT Syndrome (LQTS) is caused by mutations in a number of genes, most of which are related to the functioning of sodium or potassium ion channels in the heart.1 Testing may offer prognostic information in some cases, as specific genes and even specific mutations within those genes may have some correlation to risk for sudden death, effectiveness of beta-blocker therapy, and preventive strategies.1-4 Signs and symptoms of long QT syndrome (LQTS) are variable, but may include a prolonged QT interval on an electrocardiogram, torsades de pointes, syncope, seizures, cardiac arrest, and sudden cardiac death.1,2 Many patients with LQTS can be largely asymptomatic, with cardiac arrest or sudden cardiac death as the first and only symptom. -

Effects of Nifekalant Injected Into the Pericardial Space on the Transmural Dispersion of Repolarization in Pig
Showa Univ J Med Sci 19(3), 123 135, September 2007 Original Effects of Nifekalant Injected into the Pericardial Space on the Transmural Dispersion of Repolarization in Pig Hiroyuki ITo, Taku ASANO, Youichi KOBAYASHI, Tatsuya ONUKI, Fumito MwosHI, Taka-aki MATSUYAMA, Yoshino MINOURA, Norikazu WATANABE, Mitsuharu KAWAMURA, Kaoru TANNO and Takashl KATAGIRI Abstract: Transmural dispersion of repolarization (TDR) has been implicated in the onset of ventricular arrhythmia. We investigated the effects of nifekalant (NIF) injected into the pericardial space on TDR and T wave in pig. We injected 50 or 100 mg of NIF into the pericardial space of 11 pigs, and measured the effective refractory period (ERP) between the endocardial and epicardial myocardial cells, as well as QT time and QT peak-end as an index of TDR and T waveforms, respectively. TDR decreased from 56•} 10 msec to 44•}8 msec (P < 0.01), 5.1 min after injection of 100 mg of NIF, although the QTc did not change. At a later time, QTc increased from 457 •} 44 msec before injection to 540•}49 msec (P < 0.01) and TDR recovered to the control level. When 50 mg of NIF was injected, the T wave amplitude decreased from 0.433 •}0.301 mV before injection to 0.107•}0.192 mV (P < 0.01) at 10 min after injection ; 100 mg of NIF caused the T wave amplitude to decrease more rapidly, reaching a negative value at 4.5 min after injection. Injecting NIF into the pericardial space altered ERP, QT, and T waveform, and also decreased TDR. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627). -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Young Adults. Look for ST Elevation, Tall QRS Voltage, "Fishhook" Deformity at the J Point, and Prominent T Waves
EKG Abnormalities I. Early repolarization abnormality: A. A normal variant. Early repolarization is most often seen in healthy young adults. Look for ST elevation, tall QRS voltage, "fishhook" deformity at the J point, and prominent T waves. ST segment elevation is maximal in leads with tallest R waves. Note high take off of the ST segment in leads V4-6; the ST elevation in V2-3 is generally seen in most normal ECG's; the ST elevation in V2- 6 is concave upwards, another characteristic of this normal variant. Characteristics’ of early repolarization • notching or slurring of the terminal portion of the QRS wave • symmetric concordant T waves of large amplitude • relative temporal stability • most commonly presents in the precordial leads but often associated with it is less pronounced ST segment elevation in the limb leads To differentiate from anterior MI • the initial part of the ST segment is usually flat or convex upward in AMI • reciprocal ST depression may be present in AMI but not in early repolarization • ST segments in early repolarization are usually <2 mm (but have been reported up to 4 mm) To differentiate from pericarditis • the ST changes are more widespread in pericarditis • the T wave is normal in pericarditis • the ratio of the degree of ST elevation (measured using the PR segment as the baseline) to the height of the T wave is greater than 0.25 in V6 in pericarditis. 1 II. Acute Pericarditis: Stage 1 Pericarditis Changes A. Timing 1. Onset: Day 2-3 2. Duration: Up to 2 weeks B. Findings 1. -

Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?
The Science Journal of the Lander College of Arts and Sciences Volume 11 Number 1 Fall 2017 - 2017 Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Touro College Follow this and additional works at: https://touroscholar.touro.edu/sjlcas Part of the Cardiovascular Diseases Commons, and the Medical Genetics Commons Recommended Citation Braun, M. (2017). Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?. The Science Journal of the Lander College of Arts and Sciences, 11(1). Retrieved from https://touroscholar.touro.edu/sjlcas/vol11/iss1/8 This Article is brought to you for free and open access by the Lander College of Arts and Sciences at Touro Scholar. It has been accepted for inclusion in The Science Journal of the Lander College of Arts and Sciences by an authorized editor of Touro Scholar. For more information, please contact [email protected]. Should Genetic Testing be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Menachem Braun graduated with a BS in Biology in September 2017. Abstract 7KH/RQJ476\QGURPH /476 LVDIDPLOLDOSRWHQWLDOO\IDWDOFDUGLDFDUUK\WKPLD7UDGLWLRQDOO\LWKDVEHHQGLDJQRVHGE\(&* Molecular studies have provided evidence that LQTS can be caused by a range of underlying molecular abnormalities. Genetic research has proven that different forms of LQTS have different genotypic bases. Therefore, it has become possible to diagnose WKHVSHFLÀFW\SHRIGLVHDVHJHQHWLFDOO\7KLVVWXG\H[DPLQHVWKHDGYDQFHPHQWVPDGHLQWKHSDVWWKLUW\\HDUVLQXQGHUVWDQGLQJ /476DQGUHVHDUFKUHJDUGLQJWKHXVHRIJHQHWLFWHVWLQJLQRUGHUWRGHWHUPLQHWKHEHQHÀWVRIJHQHWLFWHVWLQJIRUWKLVGLVHDVH$ survey of original studies which produced the information is presented here, and provides the reader with an understanding of the PHFKDQLFVRIWKHGLVHDVHDQGKRZWKH\GLIIHULQWKHVHYHUDOJHQHWLFYDULDQWV5HVHDUFKVKRZVWKDWWKHEHQHÀWRIJHQHWLFWHVWLQJ must be weighed against the personal implications in may have for a particular patient and his or her family. -

Bundle Branch Blocks And/Or Hemiblocks Complicating Acute Myocardial Ischemia Or Infarction
Journal of Interventional Cardiac Electrophysiology https://doi.org/10.1007/s10840-018-0430-3 Bundle branch blocks and/or hemiblocks complicating acute myocardial ischemia or infarction Samuel Lévy1 Received: 23 May 2018 /Accepted: 24 July 2018 # Springer Science+Business Media, LLC, part of Springer Nature 2018 Abstract Despite the bulk of anatomical and histologic evidence supporting the existence of three fascicules in the left branch of the His bundle, the concept of a bifascicular system proposed by Rosenbaum and his school has been adopted by the cardiological community as a practical teaching tool. Left anterior hemiblock (LAH) refers to block of the antero-superior branch of the left branch which is small and left posterior hemiblock (LPH) to block of the postero-inferior branch which is larger. The LAH is more common that the LPH and often associated with a complete right bundle branch block (RBBB). Coronary artery disease (CAD) is a major cause of hemiblocks. In this review article, we discuss various aspects of the relation of hemiblocks with CAD. We looked at the prevalence of LAH in consecutive patients undergoing coronary angiography and who had a significant coronary lesion in one vessel or more. In all patients with LAH, a significant lesion of the left anterior descending coronary artery was present, with in the majority of patients, an impairment of the left ventricular function. Bifascicular block (RBBB with LAH or LPH) can complicate acute myocardial infarction and is often associated with a poor prognosis and the presence of heart failure. Thrombolysis and or early angioplasty in acute myocardial infarction have significantly improved the prognosis and reduced the mortality associated with bifascicular block. -

Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes
Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes Michael J. Ackerman, MD, PhD Windland Smith Rice Cardiovascular Genomics Research Professor Professor of Medicine, Pediatrics, and Pharmacology Director, Long QT Syndrome Clinic and the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory President, Sudden Arrhythmia Death Syndromes (SADS) Foundation Learning Objectives to Disclose: • To RECOGNIZE the “faces” (phenotypes) of the congenital long QT syndromes (LQTS) • To CRITIQUE the various diagnostic modalities used in the evaluation of LQTS and UNDERSTAND their limitations • To ASSESS the currently available treatment options for the various LQT syndromes and EVALUATE their efficacy WINDLAND Smith Rice Sudden Death Genomics Laboratory Conflicts of Interest to Disclose: • Consultant – Boston Scientific, Gilead Sciences, Medtronic, St. Jude Medical, and Transgenomic/FAMILION • Royalties – Transgenomic/FAMILION Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT ♥ 1957 – first clinical description – JLNS ♥ 1960s – RomanoProlonged-Ward QT syndrome ♥ 1983 – “Schwartz/Moss score”1. Syncope ♥ 1991 – first LQTS chromosome locus 2. Seizures ♥ March 10, 1995 – birth of cardiac 3. Sudden channelopathies death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal -

Genetic Testing for Hereditary Cardiac Disease
Clinical Appropriateness Guidelines Genetic Testing for Hereditary Cardiac Disease EFFECTIVE MARCH 8, 2021 Appropriate.Safe.Affordable © 2019 AIM Specialty Health 2064-0319 Table of Contents Scope .......................................................................................................................................................... 3 Genetic Counseling Requirement ............................................................................................................... 3 Appropriate Use Criteria.............................................................................................................................. 3 Confirmation/Diagnostic Testing of Affected Individuals .............................................................................. 4 Testing of Asymptomatic Individuals .............................................................................................................. 4 Post-Mortem Testing ........................................................................................................................................ 4 Long QT ............................................................................................................................................................. 5 Dilated Cardiomyopathy .................................................................................................................................. 5 Tests Not Clinically Appropriate .....................................................................................................................