Update on the Diagnosis and Management of Familial Long QT Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non Commercial Use Only
Cardiogenetics 2017; volume 7:6304 Sudden death in a young patient with atrial fibrillation Case Report Correspondence: María Angeles Espinosa Castro, Inherited Cardiovascular Disease A 22-year-old man suffered a sudden Program, Cardiology Department, Gregorio María Tamargo, cardiac arrest without previous symptoms Marañón Hospital, Dr. Esquerdo, 46, 28007, María Ángeles Espinosa, while he was at rest, waiting for a subway Madrid, Spain. Víctor Gómez-Carrillo, Miriam Juárez, train. Cardiopulmonary resuscitation was Tel.: +34.91.586.82.90. immediately started using an Automated E-mail: [email protected] Francisco Fernández-Avilés, External Defibrillation that identified the Raquel Yotti Key words: KCNQ1; mutation; channelopa- presence of ventricular fibrillation and thy; sudden cardiac death; atrial fibrillation. Inherited Cardiovascular Disease delivered a shock. Return of spontaneous Program, Cardiology Department, circulation was achieved after three Contributions: MT, acquisition and interpreta- Gregorio Marañón Hospital, Madrid, attempts, being atrial fibrillation (AF) the tion of data for the work, ensuring that ques- Spain patient’s rhythm at this point (Figure 1). tions related to the accuracy or integrity of any He was admitted to our Cardiovascular part of the work is appropriately investigated Intensive Care Unit and therapeutic and resolved; MAE, conception of the work, hypothermia was performed over a period critical revision of the intellectual content, final approval of the version to be published, Abstract of 24 h. After completing hypothermia, ensuring that questions related to the accuracy rewarming, and another 24 h of controlled of any part of the work is appropriately inves- Sudden cardiac death (SCD) in young normothermia the patient awakened with no tigated and resolved; VG-C, acquisition and patients without structural heart disease is residual neurologic damage. -

Towards Mutation-Specific Precision Medicine in Atypical Clinical
International Journal of Molecular Sciences Review Towards Mutation-Specific Precision Medicine in Atypical Clinical Phenotypes of Inherited Arrhythmia Syndromes Tadashi Nakajima * , Shuntaro Tamura, Masahiko Kurabayashi and Yoshiaki Kaneko Department of Cardiovascular Medicine, Gunma University Graduate School of Medicine, Maebashi 371-8511, Gunma, Japan; [email protected] (S.T.); [email protected] (M.K.); [email protected] (Y.K.) * Correspondence: [email protected]; Tel.: +81-27-220-8145; Fax: +81-27-220-8158 Abstract: Most causal genes for inherited arrhythmia syndromes (IASs) encode cardiac ion channel- related proteins. Genotype-phenotype studies and functional analyses of mutant genes, using heterol- ogous expression systems and animal models, have revealed the pathophysiology of IASs and enabled, in part, the establishment of causal gene-specific precision medicine. Additionally, the utilization of induced pluripotent stem cell (iPSC) technology have provided further insights into the patho- physiology of IASs and novel promising therapeutic strategies, especially in long QT syndrome. It is now known that there are atypical clinical phenotypes of IASs associated with specific mutations that have unique electrophysiological properties, which raises a possibility of mutation-specific precision medicine. In particular, patients with Brugada syndrome harboring an SCN5A R1632C mutation exhibit exercise-induced cardiac events, which may be caused by a marked activity-dependent loss of R1632C-Nav1.5 availability due to a marked delay of recovery from inactivation. This suggests that the use of isoproterenol should be avoided. Conversely, the efficacy of β-blocker needs to be examined. Patients harboring a KCND3 V392I mutation exhibit both cardiac (early repolarization syndrome and Citation: Nakajima, T.; Tamura, S.; paroxysmal atrial fibrillation) and cerebral (epilepsy) phenotypes, which may be associated with a Kurabayashi, M.; Kaneko, Y. -

Stroke Mimics and Chameleons: Quandaries in the Field
Stroke Mimics and Chameleons: Quandaries in the Field Madeleine Geraghty, MD Rockwood Multicare What’s the difference Stroke mimic: Looks like a stroke, is something else Stroke chameleon: Looks like something else, is really a stroke! Scope of the Mimic Recent eval by Briard, et al: ◦ 960 patients transported by EMS during an 18 month period ◦ 42% mimics 55% other neurologic diagnoses 20% seizures, 19% migraines, 11% peripheral neuropathies 45% non-neurologic diagnoses Cardiac 16%, psychiatric 12%, infectious 9% ◦ Neurologic mimics were younger (~64 years) than non-neurologic mimics (~70 years) Entering a new era Large vessel occlusions Now a 24 hour time window for mechanical thrombectomy ◦ Most centers will likely activate the > 6 hour patients from within the ED, still working out those details Volume of stroke mimics/chameleons in the new time window? Effects on resource management? ◦ At the hospital level? ◦ At the regional level with distance transports? Need Emergency Responder Impressions now more than ever in order to learn for the future!! General Principles Positive symptoms Indicate an excess of central nervous system neuron electrical discharges Visual: flashing lights, zig zag shapes, lines, shapes, objects sensory: paresthesia, pain motor: jerking limb movements Migraine, Seizure are characterized with having “positive” symptoms Negative symptoms Indicate a loss or reduction of central nervous system neuron function – loss of vision, hearing, sensation, limb power. TIA/Stroke present with “negative” symptoms. -
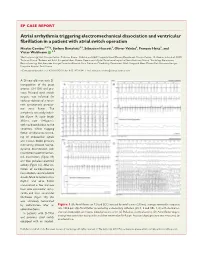
Atrial Arrhythmia Triggering Electromechanical Dissociation And
EP CASE REPORT ....................................................................................................................................................... Atrial arrhythmia triggering electromechanical dissociation and ventricular fibrillation in a patient with atrial switch operation Nicolas Combes1,2,3*, Stefano Bartoletti1,2,Se´bastien Hascoet€ 3, Olivier Vahdat2, Franc¸ois Heitz2, and Victor Waldmann 4,5 1Electrophysiology Unit, Clinique Pasteur, Toulouse, France; 2Pediatric and Adult Congenital Heart Disease Department, Clinique Pasteur, 45, Avenue de Lombez, 31076 Toulouse, France; 3Pediatric and Adult Congenital Heart Disease Department, Hoˆpital Marie Lannelongue, Le Plessis-Robinson, France; 4Cardiology Department, Electrophysiology Unit, European Georges Pompidou Hospital, Paris, France; and 5Cardiology Department, Adult Congenital Heart Disease Unit, European Georges Pompidou Hospital, Paris, France * Corresponding author. Tel: 133 562213131; fax: 133 562211641. E-mail address: [email protected] A 26-year-old man with D- transposition of the great arteries (D-TGA) and pre- vious Mustard atrial switch surgery was referred for catheter ablation of a recur- rent symptomatic paroxys- mal atrial flutter. The arrhythmia was easily induci- ble (Figure 1A, cycle length 310 ms, rate 194 b.p.m.), with rapid conduction to the ventricles. While mapping flutter, simultaneous record- ing of endocardial signals and invasive blood pressure monitoring showed haemo- dynamic deterioration with intermittent electromechan- ical dissociation (Figure 1B) and then pulseless electrical activity (Figure 1C). After ini- tiation of cardiopulmonary resuscitation, several electric shocks failed to restore sinus rhythm and atrial flutter transitioned a few minutes later into ventricular tachy- cardia and then ventricular fibrillation (Figure 1D); this was ultimately terminated by defibrillation after an Figure 1 (A) Atrial flutter on 12-lead ECG induced by atrial bursts (240 ms), average ventricular response adrenaline bolus. -

Constrictive Pericarditis Causing Ventricular Tachycardia.Pdf
EP CASE REPORT ....................................................................................................................................................... A visually striking calcific band causing monomorphic ventricular tachycardia as a first presentation of constrictive pericarditis Kian Sabzevari 1*, Eva Sammut2, and Palash Barman1 1Bristol Heart Institute, UH Bristol NHS Trust UK, UK; and 2Bristol Heart Institute, UH Bristol NHS Trust UK & University of Bristol, UK * Corresponding author. Tel: 447794900287; fax: 441173425926. E-mail address: [email protected] Introduction Constrictive pericarditis (CP) is a rare condition caused by thickening and stiffening of the pericar- dium manifesting in dia- stolic dysfunction and enhanced interventricu- lar dependence. In the developed world, most cases are idiopathic or are associated with pre- vious cardiac surgery or irradiation. Tuberculosis remains a leading cause in developing areas.1 Most commonly, CP presents with symptoms of heart failure and chest discomfort. Atrial arrhythmias have been described as a rare pre- sentation, but arrhyth- mias of ventricular origin have not been reported. Figure 1 (A) The 12 lead electrocardiogram during sustained ventricular tachycardia is shown; (B and C) Case report Different projections of three-dimensional reconstructions of cardiac computed tomography demonstrating a A 49-year-old man with a striking band of calcification around the annulus; (D) Carto 3DVR mapping—the left hand panel (i) demonstrates a background of diabetes, sinus beat with late potentials at the point of ablation in the coronary sinus, the right hand panel (iii) shows the hypertension, and hyper- pacemap with a 89% match to the clinical tachycardia [matching the morphology seen on 12 lead ECG (A)], and cholesterolaemia and a the middle panel (ii) displays the three-dimensional voltage map. -
Long QT Syndrome Testing
Lab Management Guidelines V2.0.2020 Long QT Syndrome Testing MOL.TS.196.A v2.0.2020 Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline Long QT Syndrome Sequencing 81413 Multigene Panel Long QT Syndrome Deletion/Duplication 81414 Panel Long QT Syndrome Known Familial 81403 Mutation Analysis ANK2 Sequencing 81479 CASQ2 Sequencing 81405 CAV3 Sequencing 81404 KCNE1 Sequencing 81479 KCNE2 Sequencing 81479 KCNH2 Sequencing 81406 KCNJ2 Sequencing 81403 KCNQ1 Sequencing 81406 RYR2 Sequencing 81408 SCN5A Sequencing 81407 SCN4B Sequencing 81479 AKAP9 Sequencing 81479 SNTA1 Sequencing 81479 KCNJ5 Sequencing 81479 CALM1 Sequencing 81479 CALM2 Sequencing 81479 CACNA1C Sequencing 81479 © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V2.0.2020 What is Long QT syndrome Definition Long QT Syndrome (LQTS) is caused by mutations in a number of genes, most of which are related to the functioning of sodium or potassium ion channels in the heart.1 Testing may offer prognostic information in some cases, as specific genes and even specific mutations within those genes may have some correlation to risk for sudden death, effectiveness of beta-blocker therapy, and preventive strategies.1-4 Signs and symptoms of long QT syndrome (LQTS) are variable, but may include a prolonged QT interval on an electrocardiogram, torsades de pointes, syncope, seizures, cardiac arrest, and sudden cardiac death.1,2 Many patients with LQTS can be largely asymptomatic, with cardiac arrest or sudden cardiac death as the first and only symptom. -

Cardiac Arrest: an Important Public Health Issue
Cardiac Arrest: An Important Public Health Issue Cardiac arrest is a public health issue with widespread incidence and severe impact on human health and well-being. There are several recommended strategies for prevention and control. Incidence Impact In 2015, approximately Mortality: 357,000 people experienced 70%–90% out-of-hospital cardiac arrest (OHCA) in the United Approximately 70%–90% of individuals with OHCA die States. before reaching the hospital. Approximately 209,000 Morbidity: Those who survive cardiac arrest are people are treated for in- likely to suffer from injury to the brain and hospital cardiac arrest nervous system and other physical ailments. (IHCA) each year. Additionally, nearly half of OHCA survivors suffer psychological distress such as anxiety, post traumatic stress disorder, and depression. Economic Impact Societal Cost: The estimated burden to society of death from cardiac arrest is 2 million years of life lost for men and 1.3 million years for women, greater than estimates for all individual $ cancers and most leading causes of death. Prevention Early intervention by CPR and defibrillation:Early, high-quality CPR, including compression only CPR, and use of automated external defibrillators (AEDs) immediately following cardiac arrest can reduce morbidity and save lives. Clinical prevention: For Other early interventions: Depending on the patients at high risk, cause of the cardiac arrest, other interventions implantable cardioverter such as cold therapy and administering antidote defibrillators and to toxin-related cardiac arrest can reduce pharmacologic therapies mortality and long-term side effects. can prevent cardiac arrest. What Is Public Health’s Role in Cardiac Arrest? The public health community can implement strategies to prevent and control cardiac arrest. -

Pacemaker Syndrome Pacemaker Therapy Has Become an Important Therapeutic Option for Patients with Heart Rhythm Conditions Worldwide
360 Cardiology Pacemaker syndrome Pacemaker therapy has become an important therapeutic option for patients with heart rhythm conditions worldwide. Te number of elderly patients needing pacemakers is on the increase due to an ageing population worldwide. Pacemaker syndrome consists of the cardiovascular signs and symptoms of heart failure and hypotension induced by right ventricular (RV) pacing. Dr Satnam Singh Research Registrar, University of Aberdeen, Level 3, Polwarth building, Aberdeen email [email protected] Pacemaker syndrome is a term syndrome occurring in dual trial was a single blinded study proposed in 1979 by Erbel and chamber modes.5,6 It can even enrolling around 2000 patients refers to symptoms and signs in occur with AAI pacing with long with sick sinus syndrome. the pacemaker patient caused by PR intervals. All patients were implanted inadequate timing of atrial and dual chamber pacemakers ventricular contractions.1 It was first programmed to VVIR or DDDR described in 1969 by Mitsui et al2 as Incidence before implantation. Pacemaker an iatrogenic disease characterised syndrome was a secondary by the disappearance of symptoms Te overall incidence of pacemaker endpoint studied. Severe with restoration of atrioventricular syndrome is very difficult to pacemaker syndrome developed synchrony (AV synchrony). estimate but is about 20% in a in nearly 20% of VVIR-paced It means if atria and ventricles landmark trial called the Mode patients and improved with contract at appropriate timings (as Selection Trial (MOST).7 It occurs reprogramming to the dual- close to physiological), pacemaker with equal frequency in both sexes chamber pacing mode. syndrome can be prevented. -

What Are Premature Ventricular Contractions?
What Are Premature Ventricular Contractions? Premature ventricular contractions (VPCs or PVCs) are irregular beats that originate from the heart’s pumping chambers (ventricles). These beats interrupt the normal regular (sinus) rhythm, and result in irregularity to the heart rhythm. Although single PVCs are not life- threatening, they may indicate the presence of underlying heart disease, or when they become more severe can cause rapid and dangerous increases in heart rate (ventricular tachycardia). WHAT CAUSES PREMATURE VENTRICULAR CONTRACTIONS? PVCs can occur secondary to a variety of causes, most concerning being cardiac disease. PVCs are most commonly seen in dogs with Dilated Cardiomyopathy, but can also occur in the later stages of disease with Myxomatous Mitral Valve Degeneration. PVCs and other ventricular arrhythmias can also with no evidence of underlying structural heart disease. The workup for PVCs can be frustrating, because PVCs can also occur secondary to conditions unrelated to the heart. Most commonly they can occur secondary to gastrointestinal disease, systemic disease, and pain. HOW ARE PREMATURE VENTRICULAR CONTRACTIONS DIAGNOSED? Most commonly, your veterinarian will hear an irregular heart rhythm and recommend an electrocardiogram (ECG) to assess the heart rhythm. The ECG will enable your veterinarian to determine whether the irregular rhythm is due to PVCs. Following this diagnosis, there are several additional recommended tests. TESTING Following this diagnosis, there are several additional recommended tests. ECHOCARDIOGRAPHY Echocardiography (heart ultrasound) is recommended to make sure that there is no evidence of structural cardiac disease causing the abnormal heart rhythm. 24-HOUR HOLTER Single PVCs usually do not require treatment, however a Holter monitor will determine whether the frequency or severity of the MONITOR PVCs warrant anti-arrhythmic medication, and make sure that there is no evidence of ventricular tachycardia, which is a life threatening heart rhythm. -

Syncope Fainting, Or Syncope, Is the Sudden Loss of Consciousness and Ability to Stand
Northwestern Memorial Hospital Patient Education CONDITIONS AND DISEASES Syncope Fainting, or syncope, is the sudden loss of consciousness and ability to stand. It is also called “passing out.” This common If you have any problem is the cause of many falls and injuries. One third of people faint at least once during their life. Syncope may occur questions, ask without warning or can be signaled by: ■ Feelings of weakness your physician ■ Feeling hot or sweating ■ Dizziness or nurse. ■ Visual changes ■ Nausea ■ Palpitations Causes of syncope Fainting is due to a sudden decrease in blood flow and oxygen to the brain. There are many causes of syncope, but most fall into 1 of 3 major types. Abnormal nerve reflex Nerves that control heart rate, blood pressure and other body functions may respond in an abnormal way and cause syncope. This can be triggered by: ■ Standing ■ Pain ■ Unpleasant sight or smell ■ Stress, anxiety or emotional distress ■ Coughing, sneezing or swallowing ■ Urinating or having a bowel movement Fainting due to an abnormal nerve reflex is more likely to occur under certain conditions, such as dehydration, viral infection, after prolonged bed rest or a lack of sleep or regular food intake. Types of syncope that involve an abnormal nerve reflex include: ■ Vasovagal (neurocardiac) syncope is the most common type and can occur at any age. It often occurs when blood pools in the leg veins. This triggers a reflex where the heart rate, blood pressure or both may suddenly fall. It generally is not a dangerous condition and can be prevented by avoiding situations that can trigger syncope. -

Cardiomyopathy Precision Panel Overview Indications
Cardiomyopathy Precision Panel Overview Cardiomyopathies are a group of conditions with a strong genetic background that structurally hinder the heart to pump out blood to the rest of the body due to weakness in the heart muscles. These diseases affect individuals of all ages and can lead to heart failure and sudden cardiac death. If there is a family history of cardiomyopathy it is strongly recommended to undergo genetic testing to be aware of the family risk, personal risk, and treatment options. Most types of cardiomyopathies are inherited in a dominant manner, which means that one altered copy of the gene is enough for the disease to present in an individual. The symptoms of cardiomyopathy are variable, and these diseases can present in different ways. There are 5 types of cardiomyopathies, the most common being hypertrophic cardiomyopathy: 1. Hypertrophic cardiomyopathy (HCM) 2. Dilated cardiomyopathy (DCM) 3. Restrictive cardiomyopathy (RCM) 4. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) 5. Isolated Left Ventricular Non-Compaction Cardiomyopathy (LVNC). The Igenomix Cardiomyopathy Precision Panel serves as a diagnostic and tool ultimately leading to a better management and prognosis of the disease. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes. Indications The Igenomix Cardiomyopathy Precision Panel is indicated in those cases where there is a clinical suspicion of cardiomyopathy with or without the following manifestations: - Shortness of breath - Fatigue - Arrythmia (abnormal heart rhythm) - Family history of arrhythmia - Abnormal scans - Ventricular tachycardia - Ventricular fibrillation - Chest Pain - Dizziness - Sudden cardiac death in the family 1 Clinical Utility The clinical utility of this panel is: - The genetic and molecular diagnosis for an accurate clinical diagnosis of a patient with personal or family history of cardiomyopathy, channelopathy or sudden cardiac death. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627).