Romano-Ward Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non Commercial Use Only
Cardiogenetics 2017; volume 7:6304 Sudden death in a young patient with atrial fibrillation Case Report Correspondence: María Angeles Espinosa Castro, Inherited Cardiovascular Disease A 22-year-old man suffered a sudden Program, Cardiology Department, Gregorio María Tamargo, cardiac arrest without previous symptoms Marañón Hospital, Dr. Esquerdo, 46, 28007, María Ángeles Espinosa, while he was at rest, waiting for a subway Madrid, Spain. Víctor Gómez-Carrillo, Miriam Juárez, train. Cardiopulmonary resuscitation was Tel.: +34.91.586.82.90. immediately started using an Automated E-mail: [email protected] Francisco Fernández-Avilés, External Defibrillation that identified the Raquel Yotti Key words: KCNQ1; mutation; channelopa- presence of ventricular fibrillation and thy; sudden cardiac death; atrial fibrillation. Inherited Cardiovascular Disease delivered a shock. Return of spontaneous Program, Cardiology Department, circulation was achieved after three Contributions: MT, acquisition and interpreta- Gregorio Marañón Hospital, Madrid, attempts, being atrial fibrillation (AF) the tion of data for the work, ensuring that ques- Spain patient’s rhythm at this point (Figure 1). tions related to the accuracy or integrity of any He was admitted to our Cardiovascular part of the work is appropriately investigated Intensive Care Unit and therapeutic and resolved; MAE, conception of the work, hypothermia was performed over a period critical revision of the intellectual content, final approval of the version to be published, Abstract of 24 h. After completing hypothermia, ensuring that questions related to the accuracy rewarming, and another 24 h of controlled of any part of the work is appropriately inves- Sudden cardiac death (SCD) in young normothermia the patient awakened with no tigated and resolved; VG-C, acquisition and patients without structural heart disease is residual neurologic damage. -

Screening for Copy Number Variation in Genes Associated with the Long QT Syndrome Clinical Relevance
Journal of the American College of Cardiology Vol. 57, No. 1, 2011 © 2011 by the American College of Cardiology Foundation ISSN 0735-1097/$36.00 Published by Elsevier Inc. doi:10.1016/j.jacc.2010.08.621 Heart Rhythm Disorders Screening for Copy Number Variation in Genes Associated With the Long QT Syndrome Clinical Relevance Julien Barc, PHD,*‡§ François Briec, MD,*†‡§ Sébastien Schmitt, MD,ʈ Florence Kyndt, PharmD, PHD,*‡§ʈ Martine Le Cunff, BS,*‡§ Estelle Baron, BS,*‡§ Claude Vieyres, MD,¶ Frédéric Sacher, MD,# Richard Redon, PHD,*‡§ Cédric Le Caignec, MD, PHD,*‡§ʈ Hervé Le Marec, MD, PHD,*†‡§ Vincent Probst, MD, PHD,*†‡§ Jean-Jacques Schott, PHD*†‡§ Nantes, Angoulême, and Bordeaux, France Objectives The aim of this study was to investigate, in a set of 93 mutation-negative long QT syndrome (LQTS) probands, the frequency of copy number variants (CNVs) in LQTS genes. Background LQTS is an inherited cardiac arrhythmia characterized by a prolonged heart rate–corrected QT (QTc) interval as- sociated with sudden cardiac death. Recent studies suggested the involvement of duplications or deletions in the occurrence of LQTS. However, their frequency remains unknown in LQTS patients. Methods Point mutations in KCNQ1, KCNH2, and SCN5A genes were excluded by denaturing high-performance liquid chromatography or direct sequencing. We applied Multiplex Ligation-dependent Probe Amplification (MLPA) to detect CNVs in exons of these 3 genes. Abnormal exon copy numbers were confirmed by quantitative multiplex PCR of short fluorescent fragment (QMPSF). Array-based comparative genomic hybridization (array CGH) analysis was performed using Agilent Human Genome 244K Microarrays to further map the genomic rearrangements. -

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -
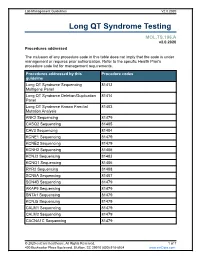
Long QT Syndrome Testing
Lab Management Guidelines V2.0.2020 Long QT Syndrome Testing MOL.TS.196.A v2.0.2020 Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline Long QT Syndrome Sequencing 81413 Multigene Panel Long QT Syndrome Deletion/Duplication 81414 Panel Long QT Syndrome Known Familial 81403 Mutation Analysis ANK2 Sequencing 81479 CASQ2 Sequencing 81405 CAV3 Sequencing 81404 KCNE1 Sequencing 81479 KCNE2 Sequencing 81479 KCNH2 Sequencing 81406 KCNJ2 Sequencing 81403 KCNQ1 Sequencing 81406 RYR2 Sequencing 81408 SCN5A Sequencing 81407 SCN4B Sequencing 81479 AKAP9 Sequencing 81479 SNTA1 Sequencing 81479 KCNJ5 Sequencing 81479 CALM1 Sequencing 81479 CALM2 Sequencing 81479 CACNA1C Sequencing 81479 © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V2.0.2020 What is Long QT syndrome Definition Long QT Syndrome (LQTS) is caused by mutations in a number of genes, most of which are related to the functioning of sodium or potassium ion channels in the heart.1 Testing may offer prognostic information in some cases, as specific genes and even specific mutations within those genes may have some correlation to risk for sudden death, effectiveness of beta-blocker therapy, and preventive strategies.1-4 Signs and symptoms of long QT syndrome (LQTS) are variable, but may include a prolonged QT interval on an electrocardiogram, torsades de pointes, syncope, seizures, cardiac arrest, and sudden cardiac death.1,2 Many patients with LQTS can be largely asymptomatic, with cardiac arrest or sudden cardiac death as the first and only symptom. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627). -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

A Study on Acute Myeloid Leukemias with Trisomy 8, 11, Or 13, Monosomy 7, Or Deletion 5Q
Leukemia (2005) 19, 1224–1228 & 2005 Nature Publishing Group All rights reserved 0887-6924/05 $30.00 www.nature.com/leu Genomic gains and losses influence expression levels of genes located within the affected regions: a study on acute myeloid leukemias with trisomy 8, 11, or 13, monosomy 7, or deletion 5q C Schoch1, A Kohlmann1, M Dugas1, W Kern1, W Hiddemann1, S Schnittger1 and T Haferlach1 1Laboratory for Leukemia Diagnostics, Department of Internal Medicine III, University Hospital Grosshadern, Ludwig-Maximilians-University, Munich, Germany We performed microarray analyses in AML with trisomies 8 aim of this study to investigate whether gains and losses on the (n ¼ 12), 11 (n ¼ 7), 13 (n ¼ 7), monosomy 7 (n ¼ 9), and deletion genomic level translate into altered genes expression also in 5q (n ¼ 7) as sole changes to investigate whether genomic gains and losses translate into altered expression levels of other areas of the genome in AML. genes located in the affected chromosomal regions. Controls were 104 AML with normal karyotype. In subgroups with trisomy, the median expression of genes located on gained Materials and methods chromosomes was higher, while in AML with monosomy 7 and deletion 5q the median expression of genes located in deleted Samples regions was lower. The 50 most differentially expressed genes, as compared to all other subtypes, were equally distributed Bone marrow samples of AML patients at diagnosis were over the genome in AML subgroups with trisomies. In contrast, 30 and 86% of the most differentially expressed genes analyzed: 12 cases with trisomy 8 (AML-TRI8), seven with characteristic for AML with 5q deletion and monosomy 7 are trisomy 11 (AML-TRI11), seven with trisomy 13 (AML-TRI13), located on chromosomes 5 or 7. -

Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?
The Science Journal of the Lander College of Arts and Sciences Volume 11 Number 1 Fall 2017 - 2017 Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Touro College Follow this and additional works at: https://touroscholar.touro.edu/sjlcas Part of the Cardiovascular Diseases Commons, and the Medical Genetics Commons Recommended Citation Braun, M. (2017). Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?. The Science Journal of the Lander College of Arts and Sciences, 11(1). Retrieved from https://touroscholar.touro.edu/sjlcas/vol11/iss1/8 This Article is brought to you for free and open access by the Lander College of Arts and Sciences at Touro Scholar. It has been accepted for inclusion in The Science Journal of the Lander College of Arts and Sciences by an authorized editor of Touro Scholar. For more information, please contact [email protected]. Should Genetic Testing be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Menachem Braun graduated with a BS in Biology in September 2017. Abstract 7KH/RQJ476\QGURPH /476 LVDIDPLOLDOSRWHQWLDOO\IDWDOFDUGLDFDUUK\WKPLD7UDGLWLRQDOO\LWKDVEHHQGLDJQRVHGE\(&* Molecular studies have provided evidence that LQTS can be caused by a range of underlying molecular abnormalities. Genetic research has proven that different forms of LQTS have different genotypic bases. Therefore, it has become possible to diagnose WKHVSHFLÀFW\SHRIGLVHDVHJHQHWLFDOO\7KLVVWXG\H[DPLQHVWKHDGYDQFHPHQWVPDGHLQWKHSDVWWKLUW\\HDUVLQXQGHUVWDQGLQJ /476DQGUHVHDUFKUHJDUGLQJWKHXVHRIJHQHWLFWHVWLQJLQRUGHUWRGHWHUPLQHWKHEHQHÀWVRIJHQHWLFWHVWLQJIRUWKLVGLVHDVH$ survey of original studies which produced the information is presented here, and provides the reader with an understanding of the PHFKDQLFVRIWKHGLVHDVHDQGKRZWKH\GLIIHULQWKHVHYHUDOJHQHWLFYDULDQWV5HVHDUFKVKRZVWKDWWKHEHQHÀWRIJHQHWLFWHVWLQJ must be weighed against the personal implications in may have for a particular patient and his or her family. -

AKAP9 Antibody (Monoclonal) (M01) Mouse Monoclonal Antibody Raised Against a Partial Recombinant AKAP9
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 AKAP9 Antibody (monoclonal) (M01) Mouse monoclonal antibody raised against a partial recombinant AKAP9. Catalog # AT1090a Specification AKAP9 Antibody (monoclonal) (M01) - Product Information Application WB, E Primary Accession Q99996 Other Accession NM_147171 Reactivity Human Host mouse Clonality Monoclonal Isotype IgG2a Kappa Calculated MW 452987 AKAP9 Antibody (monoclonal) (M01) - Additional Information Antibody Reactive Against Recombinant Protein.Western Blot detection against Gene ID 10142 Immunogen (36.74 KDa) . Other Names A-kinase anchor protein 9, AKAP-9, A-kinase anchor protein 350 kDa, AKAP 350, hgAKAP 350, A-kinase anchor protein 450 kDa, AKAP 450, AKAP 120-like protein, Centrosome- and Golgi-localized PKN-associated protein, CG-NAP, Protein hyperion, Protein kinase A-anchoring protein 9, PRKA9, Protein yotiao, AKAP9, AKAP350, AKAP450, KIAA0803 Target/Specificity Detection limit for recombinant GST tagged AKAP9 (NP_671700, 3812 a.a. ~ 3911 a.a) AKAP9 is approximately 0.3ng/ml as a partial recombinant protein with GST tag. capture antibody. MW of the GST tag alone is 26 KDa. Dilution AKAP9 Antibody (monoclonal) (M01) - WB~~1:500~1000 Background Format The A-kinase anchor proteins (AKAPs) are a Clear, colorless solution in phosphate group of structurally diverse proteins which buffered saline, pH 7.2 . have the common function of binding to the regulatory subunit of protein kinase A (PKA) Storage and confining the holoenzyme to discrete Store at -20°C or lower. Aliquot to avoid locations within the cell. This gene encodes a repeated freezing and thawing. member of the AKAP family. -

Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes
Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes Michael J. Ackerman, MD, PhD Windland Smith Rice Cardiovascular Genomics Research Professor Professor of Medicine, Pediatrics, and Pharmacology Director, Long QT Syndrome Clinic and the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory President, Sudden Arrhythmia Death Syndromes (SADS) Foundation Learning Objectives to Disclose: • To RECOGNIZE the “faces” (phenotypes) of the congenital long QT syndromes (LQTS) • To CRITIQUE the various diagnostic modalities used in the evaluation of LQTS and UNDERSTAND their limitations • To ASSESS the currently available treatment options for the various LQT syndromes and EVALUATE their efficacy WINDLAND Smith Rice Sudden Death Genomics Laboratory Conflicts of Interest to Disclose: • Consultant – Boston Scientific, Gilead Sciences, Medtronic, St. Jude Medical, and Transgenomic/FAMILION • Royalties – Transgenomic/FAMILION Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT ♥ 1957 – first clinical description – JLNS ♥ 1960s – RomanoProlonged-Ward QT syndrome ♥ 1983 – “Schwartz/Moss score”1. Syncope ♥ 1991 – first LQTS chromosome locus 2. Seizures ♥ March 10, 1995 – birth of cardiac 3. Sudden channelopathies death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal -

Novel AKAP9 Mutation and Long QT Syndrome in a Patient with Torsades Des Pointes
Journal of Interventional Cardiac Electrophysiology (2019) 56:171–172 https://doi.org/10.1007/s10840-019-00606-y CASE REPORTS Novel AKAP9 mutation and long QT syndrome in a patient with torsades des pointes Dario Bottigliero1 & Ilenia Monaco1 & Rosa Santacroce1 & Grazia Casavecchia1 & Michele Correale1 & Francesca Guastafierro1 & Angelica Leccese1 & Giorgia Cordisco1 & Riccardo Ieva2 & Roberta Trunzo1 & Matteo Di Biase3 & Maurizio Margaglione1 & Natale Daniele Brunetti1 Received: 22 March 2019 /Accepted: 2 August 2019 /Published online: 15 August 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 We report the case of an 84-year-old man with hyper- DNA: heterozygosity for AKAP9 exon 9 (c.3673C>T tension, diabetes, dyslipidemia, paroxysmal atrial fibril- G>A, p.Leu1150Phe) and mutation (Fig. 1). The vari- lation, CABG, previous right nephrectomy, and post- ants have not been previously described in the literature surgical hypothyroidism, who was admitted to neurosur- and have not been previously reported in the Human gery for subdural hematoma after syncope. Admission Gene Mutation Database (HGMD); no other possible electrocardiogram showed sinus bradycardia with causative mutations were found. prolonged QT duration. Echocardiography showed left The in silico analysis performed using SIFT and PolyPhen ventricular hypertrophy with severe mitral regurgitation. modeling programs suggests that this new variant may be During hospitalization, several episodes of torsade de harmful. pointes (8 s longest duration), treated with medical ther- AKAP9 gene is a known modifier of LQTS clinical phe- apy (magnesium sulfate iv), occurred. ECG monitoring notype by altering QTc duration and influencing the risk of constantly showed long QT (QTC > 570–580 ms) while cardiac events and the severity of the disease. -

Whole Transcriptomic Expression Differences in EBV Immortalized Versus Primary B-Cells
W&M ScholarWorks Undergraduate Honors Theses Theses, Dissertations, & Master Projects 12-2010 Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells Dolores Huberts College of William and Mary Follow this and additional works at: https://scholarworks.wm.edu/honorstheses Part of the Biology Commons Recommended Citation Huberts, Dolores, "Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B- Cells" (2010). Undergraduate Honors Theses. Paper 347. https://scholarworks.wm.edu/honorstheses/347 This Honors Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Undergraduate Honors Theses by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells A thesis submitted in partial fulfillment of the requirement for the degree of Bachelor of Science with Honors in Biology from the College of William and Mary in Virginia By Dolores Huberts Accepted for Honors ________________________________________ Lizabeth A. Allison, Director ________________________________________ Matthew Wawersik ________________________________________ Drew LaMar ________________________________________ Beverly Sher Williamsburg, Virginia December 17, 2010 ABSTRACT The Epstein–Barr Virus (EBV) is a human gamma herpes virus that infects more than 90% of the human population worldwide. It is commonly known in the US as the cause of Infectious Mononucleosis, and around the world as the cause of nasopharyngeal carcinoma and malignant lymphomas such as non-Hodgkin lymphoma, endemic Burkett’s lymphoma and Hodgkin lymphoma. Additionally, the EBV is used to immortalize cells to create cell lines for in-vitro studies.