Screening for Copy Number Variation in Genes Associated with the Long QT Syndrome Clinical Relevance
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -

A Study on Acute Myeloid Leukemias with Trisomy 8, 11, Or 13, Monosomy 7, Or Deletion 5Q
Leukemia (2005) 19, 1224–1228 & 2005 Nature Publishing Group All rights reserved 0887-6924/05 $30.00 www.nature.com/leu Genomic gains and losses influence expression levels of genes located within the affected regions: a study on acute myeloid leukemias with trisomy 8, 11, or 13, monosomy 7, or deletion 5q C Schoch1, A Kohlmann1, M Dugas1, W Kern1, W Hiddemann1, S Schnittger1 and T Haferlach1 1Laboratory for Leukemia Diagnostics, Department of Internal Medicine III, University Hospital Grosshadern, Ludwig-Maximilians-University, Munich, Germany We performed microarray analyses in AML with trisomies 8 aim of this study to investigate whether gains and losses on the (n ¼ 12), 11 (n ¼ 7), 13 (n ¼ 7), monosomy 7 (n ¼ 9), and deletion genomic level translate into altered genes expression also in 5q (n ¼ 7) as sole changes to investigate whether genomic gains and losses translate into altered expression levels of other areas of the genome in AML. genes located in the affected chromosomal regions. Controls were 104 AML with normal karyotype. In subgroups with trisomy, the median expression of genes located on gained Materials and methods chromosomes was higher, while in AML with monosomy 7 and deletion 5q the median expression of genes located in deleted Samples regions was lower. The 50 most differentially expressed genes, as compared to all other subtypes, were equally distributed Bone marrow samples of AML patients at diagnosis were over the genome in AML subgroups with trisomies. In contrast, 30 and 86% of the most differentially expressed genes analyzed: 12 cases with trisomy 8 (AML-TRI8), seven with characteristic for AML with 5q deletion and monosomy 7 are trisomy 11 (AML-TRI11), seven with trisomy 13 (AML-TRI13), located on chromosomes 5 or 7. -

AKAP9 Antibody (Monoclonal) (M01) Mouse Monoclonal Antibody Raised Against a Partial Recombinant AKAP9
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 AKAP9 Antibody (monoclonal) (M01) Mouse monoclonal antibody raised against a partial recombinant AKAP9. Catalog # AT1090a Specification AKAP9 Antibody (monoclonal) (M01) - Product Information Application WB, E Primary Accession Q99996 Other Accession NM_147171 Reactivity Human Host mouse Clonality Monoclonal Isotype IgG2a Kappa Calculated MW 452987 AKAP9 Antibody (monoclonal) (M01) - Additional Information Antibody Reactive Against Recombinant Protein.Western Blot detection against Gene ID 10142 Immunogen (36.74 KDa) . Other Names A-kinase anchor protein 9, AKAP-9, A-kinase anchor protein 350 kDa, AKAP 350, hgAKAP 350, A-kinase anchor protein 450 kDa, AKAP 450, AKAP 120-like protein, Centrosome- and Golgi-localized PKN-associated protein, CG-NAP, Protein hyperion, Protein kinase A-anchoring protein 9, PRKA9, Protein yotiao, AKAP9, AKAP350, AKAP450, KIAA0803 Target/Specificity Detection limit for recombinant GST tagged AKAP9 (NP_671700, 3812 a.a. ~ 3911 a.a) AKAP9 is approximately 0.3ng/ml as a partial recombinant protein with GST tag. capture antibody. MW of the GST tag alone is 26 KDa. Dilution AKAP9 Antibody (monoclonal) (M01) - WB~~1:500~1000 Background Format The A-kinase anchor proteins (AKAPs) are a Clear, colorless solution in phosphate group of structurally diverse proteins which buffered saline, pH 7.2 . have the common function of binding to the regulatory subunit of protein kinase A (PKA) Storage and confining the holoenzyme to discrete Store at -20°C or lower. Aliquot to avoid locations within the cell. This gene encodes a repeated freezing and thawing. member of the AKAP family. -

Novel AKAP9 Mutation and Long QT Syndrome in a Patient with Torsades Des Pointes
Journal of Interventional Cardiac Electrophysiology (2019) 56:171–172 https://doi.org/10.1007/s10840-019-00606-y CASE REPORTS Novel AKAP9 mutation and long QT syndrome in a patient with torsades des pointes Dario Bottigliero1 & Ilenia Monaco1 & Rosa Santacroce1 & Grazia Casavecchia1 & Michele Correale1 & Francesca Guastafierro1 & Angelica Leccese1 & Giorgia Cordisco1 & Riccardo Ieva2 & Roberta Trunzo1 & Matteo Di Biase3 & Maurizio Margaglione1 & Natale Daniele Brunetti1 Received: 22 March 2019 /Accepted: 2 August 2019 /Published online: 15 August 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 We report the case of an 84-year-old man with hyper- DNA: heterozygosity for AKAP9 exon 9 (c.3673C>T tension, diabetes, dyslipidemia, paroxysmal atrial fibril- G>A, p.Leu1150Phe) and mutation (Fig. 1). The vari- lation, CABG, previous right nephrectomy, and post- ants have not been previously described in the literature surgical hypothyroidism, who was admitted to neurosur- and have not been previously reported in the Human gery for subdural hematoma after syncope. Admission Gene Mutation Database (HGMD); no other possible electrocardiogram showed sinus bradycardia with causative mutations were found. prolonged QT duration. Echocardiography showed left The in silico analysis performed using SIFT and PolyPhen ventricular hypertrophy with severe mitral regurgitation. modeling programs suggests that this new variant may be During hospitalization, several episodes of torsade de harmful. pointes (8 s longest duration), treated with medical ther- AKAP9 gene is a known modifier of LQTS clinical phe- apy (magnesium sulfate iv), occurred. ECG monitoring notype by altering QTc duration and influencing the risk of constantly showed long QT (QTC > 570–580 ms) while cardiac events and the severity of the disease. -

Whole Transcriptomic Expression Differences in EBV Immortalized Versus Primary B-Cells
W&M ScholarWorks Undergraduate Honors Theses Theses, Dissertations, & Master Projects 12-2010 Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells Dolores Huberts College of William and Mary Follow this and additional works at: https://scholarworks.wm.edu/honorstheses Part of the Biology Commons Recommended Citation Huberts, Dolores, "Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B- Cells" (2010). Undergraduate Honors Theses. Paper 347. https://scholarworks.wm.edu/honorstheses/347 This Honors Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Undergraduate Honors Theses by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells A thesis submitted in partial fulfillment of the requirement for the degree of Bachelor of Science with Honors in Biology from the College of William and Mary in Virginia By Dolores Huberts Accepted for Honors ________________________________________ Lizabeth A. Allison, Director ________________________________________ Matthew Wawersik ________________________________________ Drew LaMar ________________________________________ Beverly Sher Williamsburg, Virginia December 17, 2010 ABSTRACT The Epstein–Barr Virus (EBV) is a human gamma herpes virus that infects more than 90% of the human population worldwide. It is commonly known in the US as the cause of Infectious Mononucleosis, and around the world as the cause of nasopharyngeal carcinoma and malignant lymphomas such as non-Hodgkin lymphoma, endemic Burkett’s lymphoma and Hodgkin lymphoma. Additionally, the EBV is used to immortalize cells to create cell lines for in-vitro studies. -

Myogenin Controls Via AKAP6 Non-Centrosomal Microtubule
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.11.421263; this version posted December 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Myogenin controls via AKAP6 non-centrosomal microtubule 2 organizing center formation at the nuclear envelope 3 4 Robert Becker1, Silvia Vergarajauregui1, Florian Billing1, Maria Sharkova1, 5 Eleonora Lippolis2, Kamel Mamchaoui3, Fulvia Ferrazzi2,4,5,6, Felix B. Engel1,6,* 6 7 1Experimental Renal and Cardiovascular Research, Department of Nephropathology, 8 Institute of Pathology, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), 9 91054 Erlangen, Germany 10 2Institute of Human Genetics, Friedrich-Alexander-Universität Erlangen-Nürnberg, 11 91054 Erlangen, Germany 12 3Sorbonne Universités UPMC Université Paris 06, INSERM U974, CNRS FRE3617, 13 Center for Research in Myology, GH Pitié Salpêtrière, 47 Boulevard de l’Hôpital, 75013 14 Paris, France 15 4Department of Nephropathology, Institute of Pathology, Friedrich-Alexander- 16 Universität Erlangen-Nürnberg (FAU), 91054 Erlangen, Germany 17 5Institute of Pathology, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), 18 91054 Erlangen, Germany 19 6Muscle Research Center Erlangen (MURCE), 91054 Erlangen, Germany 20 21 Short title: Myogenin controls ncMTOC formation 22 23 *Corresponding author: 24 Felix B. Engel, Universitätsklinikum Erlangen, Schwabachanlage 12 (TRC), 91054 25 Erlangen, Germany, [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.12.11.421263; this version posted December 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. -
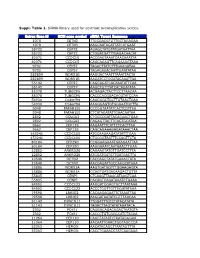
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

BRAF Fusions Define a Distinct Molecular Subset of Melanomas with Potential Sensitivity to MEK Inhibition
Clinical Cancer Human Cancer Biology Research BRAF Fusions Define a Distinct Molecular Subset of Melanomas with Potential Sensitivity to MEK Inhibition Katherine E. Hutchinson1, Doron Lipson6, Philip J. Stephens6, Geoff Otto6, Brian D. Lehmann2, Pamela L. Lyle3, Cindy L. Vnencak-Jones3,5, Jeffrey S. Ross6,7, Jennifer A. Pietenpol2, Jeffrey A. Sosman4, Igor Puzanov4, Vincent A. Miller6, and William Pao1,3,4 Abstract Purpose: Recurrent "driver" mutations at specific loci in BRAF, NRAS, KIT, GNAQ, and GNA11 define clinically relevant molecular subsets of melanoma, but more than 30% are "pan-negative" for these recurrent mutations. We sought to identify additional potential drivers in "pan-negative" melanoma. Experimental Design: Using a targeted next-generation sequencing (NGS) assay (FoundationOneÔ) and targeted RNA sequencing, we identified a novel PAPSS1-BRAF fusion in a "pan-negative" melanoma. We then analyzed NGS data from 51 additional melanomas genotyped by FoundationOneÔ, as well as melanoma RNA, whole-genome and whole-exome sequencing data in The Cancer Genome Atlas (TCGA), to determine the potential frequency of BRAF fusions in melanoma. We characterized the signaling properties of confirmed molecular alterations by ectopic expression of engineered cDNAs in 293H cells. Results: Activation of the mitogen-activated protein kinase (MAPK) pathway in cells by ectopic expression of PAPSS1-BRAF was abrogated by mitogen-activated protein kinase kinase (MEK) inhibition but not by BRAF inhibition. NGS data analysis of 51 additional melanomas revealed a second BRAF fusion (TRIM24-BRAF) in a "pan-negative" sample; MAPK signaling induced by TRIM24-BRAF was also MEK inhibitor sensitive. Through mining TCGA skin cutaneous melanoma dataset, we further identified two potential BRAF fusions in another 49 "pan-negative" cases. -

Mutation of an A-Kinase-Anchoring Protein Causes Long-QT Syndrome
Mutation of an A-kinase-anchoring protein causes long-QT syndrome Lei Chen*, Michelle L. Marquardt†, David J. Tester†, Kevin J. Sampson*, Michael J. Ackerman†‡, and Robert S. Kass*§ *Department of Pharmacology, College of Physicians and Surgeons of Columbia University, New York, NY 10032; and †Departments of Medicine, Pediatrics, and Molecular Pharmacology and Therapeutics, Divisions of Cardiovascular Diseases and Pediatric Cardiology, Mayo Clinic College of Medicine, Rochester, MN 55905 Communicated by Andrew R. Marks, Columbia University College of Physicians and Surgeons, New York, NY, November 5, 2007 (received for review August 28, 2007) A-kinase anchoring proteins (AKAPs) recruit signaling molecules pathway in the human heart. Variants of LQTS have been shown and present them to downstream targets to achieve efficient to be caused by mutations in the IKs channel ␣ (KCNQ1, LQT1) spatial and temporal control of their phosphorylation state. In the (10) or  (KCNE1, LQT5) (11) subunits. Furthermore, the heart, sympathetic nervous system (SNS) regulation of cardiac physiologic response of the heart to sympathetic nerve stimula-  action potential duration (APD), mediated by -adrenergic recep- tion (SNS) requires PKA-dependent phosphorylation of the IKs tor (AR) activation, requires assembly of AKAP9 (Yotiao) with the channel, mediated in turn by the binding of Yotiao to the IKs potassium channel ␣ subunit (KCNQ1). KCNQ1 mutations that KCNQ1 carboxyl terminal (C-T) domain (7, 12). Patients with disrupt this complex cause type 1 long-QT syndrome (LQT1), one of LQT1-causing mutations are vulnerable to LQT-precipitated the potentially lethal heritable arrhythmia syndromes. Here, we cardiac events during exertion when SNS activity is increased report identification of (i) regions on Yotiao critical to its binding (13). -

Cardiovascular Diseases Genetic Testing Program Information
Cardiovascular Diseases Genetic Testing Program Description: Congenital Heart Disease Panels We offer comprehensive gene panels designed to • Congenital Heart Disease Panel (187 genes) diagnose the most common genetic causes of hereditary • Heterotaxy Panel (114 genes) cardiovascular diseases. Testing is available for congenital • RASopathy/Noonan Spectrum Disorders Panel heart malformation, cardiomyopathy, arrythmia, thoracic (31 genes) aortic aneurysm, pulmonary arterial hypertension, Marfan Other Panels syndrome, and RASopathy/Noonan spectrum disorders. • Pulmonary Arterial Hypertension (PAH) Panel Hereditary cardiovascular disease is caused by variants in (20 genes) many different genes, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Other Indications: than condition-specific panels, we also offer single gene Panels: sequencing for any gene on the panels, targeted variant • Confirmation of genetic diagnosis in a patient with analysis, and targeted deletion/duplication analysis. a clinical diagnosis of cardiovascular disease Tests Offered: • Carrier or pre-symptomatic diagnosis identification Arrythmia Panels in individuals with a family history of cardiovascular • Comprehensive Arrhythmia Panel (81 genes) disease of unknown genetic basis • Atrial Fibrillation (A Fib) Panel (28 genes) Gene Specific Sequencing: • Atrioventricular Block (AV Block) Panel (7 genes) • Confirmation of genetic diagnosis in a patient with • Brugada Syndrome Panel (21 genes) cardiovascular disease and in whom a specific -

Romano-Ward Syndrome
Romano-Ward syndrome Description Romano-Ward syndrome is a condition that causes a disruption of the heart's normal rhythm (arrhythmia). This disorder is a form of long QT syndrome, which is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. The term "long QT" refers to a specific pattern of heart activity that is detected with an electrocardiogram (ECG or EKG), which is a test used to measure the electrical activity of the heart. In people with long QT syndrome, the part of the heartbeat known as the QT interval is abnormally long. Abnormalities in the time it takes to recharge the heart lead to abnormal heart rhythms. The arrhythmia associated with Romano-Ward syndrome can lead to fainting (syncope) or cardiac arrest and sudden death. However, some people with Romano-Ward syndrome never experience any health problems associated with the condition. Fifteen types of long QT syndrome have been defined based on their genetic cause. Some types of long QT syndrome involve other cardiac abnormalities or problems with additional body systems. Romano-Ward syndrome encompasses those types that involve only a long QT interval without other abnormalities. Frequency Romano-Ward syndrome is the most common form of inherited long QT syndrome, which affects an estimated 1 in 2,000 people worldwide. Long QT syndrome may actually be more common than this estimate, however, because some people never experience any symptoms associated with arrhythmia and therefore may not be diagnosed. Causes Mutations in the KCNQ1, KCNH2, and SCN5A genes are the most common causes of Romano-Ward syndrome. -

Anti-Cip4 Antibody (ARG42789)
Product datasheet [email protected] ARG42789 Package: 100 μl anti-Cip4 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes Cip4 Tested Reactivity Hu Tested Application WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name Cip4 Antigen Species Human Immunogen Synthetic peptide of Human Cip4. Conjugation Un-conjugated Alternate Names CIP4; Cdc42-interacting protein 4; Salt tolerant protein; STOT; HSTP; Thyroid receptor-interacting protein 10; STP; TR-interacting protein 10; hSTP; Protein Felic; TRIP-10 Application Instructions Application table Application Dilution WB 1:1000 - 1:5000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control K562 Calculated Mw 68 kDa Observed Size ~ 85 kDa Properties Form Liquid Purification Affinity purified. Buffer 50 nM Tris-Glycine (pH 7.4), 0.15 M NaCl, 0.01% Sodium azide, 40% Glycerol and 0.05% BSA. Preservative 0.01% Sodium azide Stabilizer 40% Glycerol and 0.05% BSA Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol TRIP10 Gene Full Name thyroid hormone receptor interactor 10 Function Required for translocation of GLUT4 to the plasma membrane in response to insulin signaling (By similarity).