Please Find the Updated Gene List Here
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Centronuclear Myopathies Under Attack: a Plethora of Therapeutic Targets Hichem Tasfaout, Belinda Cowling, Jocelyn Laporte
CORE Metadata, citation and similar papers at core.ac.uk Provided by Archive Ouverte en Sciences de l'Information et de la Communication Centronuclear myopathies under attack: A plethora of therapeutic targets Hichem Tasfaout, Belinda Cowling, Jocelyn Laporte To cite this version: Hichem Tasfaout, Belinda Cowling, Jocelyn Laporte. Centronuclear myopathies under attack: A plethora of therapeutic targets. Journal of Neuromuscular Diseases, IOS Press, 2018, 5, pp.387 - 406. 10.3233/JND-180309. hal-02438924 HAL Id: hal-02438924 https://hal.archives-ouvertes.fr/hal-02438924 Submitted on 14 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Journal of Neuromuscular Diseases 5 (2018) 387–406 387 DOI 10.3233/JND-180309 IOS Press Review Centronuclear myopathies under attack: A plethora of therapeutic targets Hichem Tasfaouta,b,c,d, Belinda S. Cowlinga,b,c,d,1 and Jocelyn Laportea,b,c,d,1,∗ aDepartment of Translational Medicine and Neurogenetics, Institut de G´en´etique et de Biologie Mol´eculaire et Cellulaire (IGBMC), Illkirch, France bInstitut National de la Sant´eetdelaRechercheM´edicale (INSERM), U1258, Illkirch, France cCentre National de la Recherche Scientifique (CNRS), UMR7104, Illkirch, France dUniversit´e de Strasbourg, Illkirch, France Abstract. -

Non Commercial Use Only
Cardiogenetics 2017; volume 7:6304 Sudden death in a young patient with atrial fibrillation Case Report Correspondence: María Angeles Espinosa Castro, Inherited Cardiovascular Disease A 22-year-old man suffered a sudden Program, Cardiology Department, Gregorio María Tamargo, cardiac arrest without previous symptoms Marañón Hospital, Dr. Esquerdo, 46, 28007, María Ángeles Espinosa, while he was at rest, waiting for a subway Madrid, Spain. Víctor Gómez-Carrillo, Miriam Juárez, train. Cardiopulmonary resuscitation was Tel.: +34.91.586.82.90. immediately started using an Automated E-mail: [email protected] Francisco Fernández-Avilés, External Defibrillation that identified the Raquel Yotti Key words: KCNQ1; mutation; channelopa- presence of ventricular fibrillation and thy; sudden cardiac death; atrial fibrillation. Inherited Cardiovascular Disease delivered a shock. Return of spontaneous Program, Cardiology Department, circulation was achieved after three Contributions: MT, acquisition and interpreta- Gregorio Marañón Hospital, Madrid, attempts, being atrial fibrillation (AF) the tion of data for the work, ensuring that ques- Spain patient’s rhythm at this point (Figure 1). tions related to the accuracy or integrity of any He was admitted to our Cardiovascular part of the work is appropriately investigated Intensive Care Unit and therapeutic and resolved; MAE, conception of the work, hypothermia was performed over a period critical revision of the intellectual content, final approval of the version to be published, Abstract of 24 h. After completing hypothermia, ensuring that questions related to the accuracy rewarming, and another 24 h of controlled of any part of the work is appropriately inves- Sudden cardiac death (SCD) in young normothermia the patient awakened with no tigated and resolved; VG-C, acquisition and patients without structural heart disease is residual neurologic damage. -

The Role of Z-Disc Proteins in Myopathy and Cardiomyopathy
International Journal of Molecular Sciences Review The Role of Z-disc Proteins in Myopathy and Cardiomyopathy Kirsty Wadmore 1,†, Amar J. Azad 1,† and Katja Gehmlich 1,2,* 1 Institute of Cardiovascular Sciences, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT, UK; [email protected] (K.W.); [email protected] (A.J.A.) 2 Division of Cardiovascular Medicine, Radcliffe Department of Medicine and British Heart Foundation Centre of Research Excellence Oxford, University of Oxford, Oxford OX3 9DU, UK * Correspondence: [email protected]; Tel.: +44-121-414-8259 † These authors contributed equally. Abstract: The Z-disc acts as a protein-rich structure to tether thin filament in the contractile units, the sarcomeres, of striated muscle cells. Proteins found in the Z-disc are integral for maintaining the architecture of the sarcomere. They also enable it to function as a (bio-mechanical) signalling hub. Numerous proteins interact in the Z-disc to facilitate force transduction and intracellular signalling in both cardiac and skeletal muscle. This review will focus on six key Z-disc proteins: α-actinin 2, filamin C, myopalladin, myotilin, telethonin and Z-disc alternatively spliced PDZ-motif (ZASP), which have all been linked to myopathies and cardiomyopathies. We will summarise pathogenic variants identified in the six genes coding for these proteins and look at their involvement in myopathy and cardiomyopathy. Listing the Minor Allele Frequency (MAF) of these variants in the Genome Aggregation Database (GnomAD) version 3.1 will help to critically re-evaluate pathogenicity based on variant frequency in normal population cohorts. -

Affected Female Carriers of MTM1 Mutations Display a Wide Spectrum
Acta Neuropathol DOI 10.1007/s00401-017-1748-0 ORIGINAL PAPER Afected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: delineating diagnostic clues Valérie Biancalana1,2,3,4,5 · Sophie Scheidecker1 · Marguerite Miguet1 · Annie Laquerrière6 · Norma B. Romero7,8 · Tanya Stojkovic8 · Osorio Abath Neto9 · Sandra Mercier10,11,12 · Nicol Voermans13 · Laura Tanner14 · Curtis Rogers15 · Elisabeth Ollagnon‑Roman16 · Helen Roper17 · Célia Boutte18 · Shay Ben‑Shachar19 · Xavière Lornage2,3,4,5 · Nasim Vasli2,3,4,5 · Elise Schaefer20 · Pascal Laforet21 · Jean Pouget22 · Alexandre Moerman23 · Laurent Pasquier24 · Pascale Marcorelle25,26 · Armelle Magot12 · Benno Küsters27 · Nathalie Streichenberger28 · Christine Tranchant29 · Nicolas Dondaine1 · Raphael Schneider2,3,4,5,30 · Claire Gasnier1 · Nadège Calmels1 · Valérie Kremer31 · Karine Nguyen32 · Julie Perrier12 · Erik Jan Kamsteeg33 · Pierre Carlier34 · Robert‑Yves Carlier35 · Julie Thompson30 · Anne Boland36 · Jean‑François Deleuze36 · Michel Fardeau7,8 · Edmar Zanoteli9 · Bruno Eymard21 · Jocelyn Laporte2,3,4,5 Received: 9 May 2017 / Revised: 24 June 2017 / Accepted: 2 July 2017 © Springer-Verlag GmbH Germany 2017 Abstract X-linked myotubular myopathy (XLMTM), a females and to delineate diagnostic clues, we character- severe congenital myopathy, is caused by mutations in the ized 17 new unrelated afected females and performed a MTM1 gene located on the X chromosome. A majority of detailed comparison with previously reported cases at the afected males die in the early postnatal period, whereas clinical, muscle imaging, histological, ultrastructural and female carriers are believed to be usually asymptomatic. molecular levels. Taken together, the analysis of this large Nevertheless, several afected females have been reported. cohort of 43 cases highlights a wide spectrum of clini- To assess the phenotypic and pathological spectra of carrier cal severity ranging from severe neonatal and generalized weakness, similar to XLMTM male, to milder adult forms. -

A Mutation in Lamin A/C Gene Previously Known to Cause Emery
ical C lin as C e f R o l e a p n o r r t u s o J Journal of Clinical Case Reports Chalissery et al., J Clin Case Rep 2016, 6:4 ISSN: 2165-7920 DOI: 10.4172/2165-7920.1000770 Case Report Open Access A Mutation in Lamin A/C Gene Previously Known to Cause Emery- Driefuss Muscular Dystrophy Causing A Phenotype of Limb Girdle Muscular Dystrophy Type 1B Albi J Chalissery1*, Tudor Munteanu1, Yvonne Langan2, Francesca Brett2 and Janice Redmond1 1Department of Neurology, St James’s Hospital, Ireland 2Department of Neurophysiology, St James’s Hospital, Ireland 3Department of Neuropathology, Beaumont Hospital, Dublin, Ireland *Corresponding author: Albi J Chalissery, Department of Neurology, St James’s Hospital, James’s Street, Dublin 8, Ireland, Tel +353 1 410 3000; E-mail: [email protected] Rec date: Feb 19, 2016; Acc date: Apr 13, 2016; Pub date: Apr 18, 2016 Copyright: © 2016 Chalissery AJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Mutations in the lamin protein(found in the nuclear envelope) known to cause different allelic disorders including limb girdle muscular dystrophies (LGMD) and Emery-Dreifuss muscular dystrophy (EDMD). LGMDs are a heterogeneous group of disorders with progressive proximal muscle weakness in an autosomal inheritance pattern. LGMD type 1B is a disorder secondary to a mutation in the gene encoding Lamin A/C protein in the nuclear envelope. -

Cover Petra B
UvA-DARE (Digital Academic Repository) The role of Polycomb group proteins throughout development : f(l)avoring repression van der Stoop, P.M. Link to publication Citation for published version (APA): van der Stoop, P. M. (2009). The role of Polycomb group proteins throughout development : f(l)avoring repression. Amsterdam: Nederlands Kanker Instituut - Antoni Van Leeuwenhoekziekenhuis. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (http://dare.uva.nl) Download date: 27 Oct 2019 Chapter 4 Ubiquitin E3 Ligase Ring1b/Rnf2 of Polycomb Repressive Complex 1 Contributes to Stable Maintenance of Mouse Embryonic Stem Cells Petra van der Stoop*, Erwin A. Boutsma*, Danielle Hulsman, Sonja Noback, Mike Heimerikx, Ron M. Kerkhoven, J. Willem Voncken, Lodewyk F.A. Wessels, Maarten van Lohuizen * These authors contributed equally to this work Adapted from: PLoS ONE (2008) 3(5): e2235 Ring1b regulates ES cell fate Ubiquitin E3 Ligase Ring1b/Rnf2 of Polycomb Repressive Complex 1 Contributes to Stable Maintenance of Mouse Embryonic Stem Cells Petra van der Stoop1*, Erwin A. -
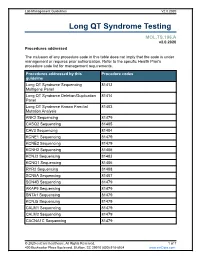
Long QT Syndrome Testing
Lab Management Guidelines V2.0.2020 Long QT Syndrome Testing MOL.TS.196.A v2.0.2020 Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline Long QT Syndrome Sequencing 81413 Multigene Panel Long QT Syndrome Deletion/Duplication 81414 Panel Long QT Syndrome Known Familial 81403 Mutation Analysis ANK2 Sequencing 81479 CASQ2 Sequencing 81405 CAV3 Sequencing 81404 KCNE1 Sequencing 81479 KCNE2 Sequencing 81479 KCNH2 Sequencing 81406 KCNJ2 Sequencing 81403 KCNQ1 Sequencing 81406 RYR2 Sequencing 81408 SCN5A Sequencing 81407 SCN4B Sequencing 81479 AKAP9 Sequencing 81479 SNTA1 Sequencing 81479 KCNJ5 Sequencing 81479 CALM1 Sequencing 81479 CALM2 Sequencing 81479 CACNA1C Sequencing 81479 © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V2.0.2020 What is Long QT syndrome Definition Long QT Syndrome (LQTS) is caused by mutations in a number of genes, most of which are related to the functioning of sodium or potassium ion channels in the heart.1 Testing may offer prognostic information in some cases, as specific genes and even specific mutations within those genes may have some correlation to risk for sudden death, effectiveness of beta-blocker therapy, and preventive strategies.1-4 Signs and symptoms of long QT syndrome (LQTS) are variable, but may include a prolonged QT interval on an electrocardiogram, torsades de pointes, syncope, seizures, cardiac arrest, and sudden cardiac death.1,2 Many patients with LQTS can be largely asymptomatic, with cardiac arrest or sudden cardiac death as the first and only symptom. -

Neuromuscular Disorders Neurology in Practice: Series Editors: Robert A
Neuromuscular Disorders neurology in practice: series editors: robert a. gross, department of neurology, university of rochester medical center, rochester, ny, usa jonathan w. mink, department of neurology, university of rochester medical center,rochester, ny, usa Neuromuscular Disorders edited by Rabi N. Tawil, MD Professor of Neurology University of Rochester Medical Center Rochester, NY, USA Shannon Venance, MD, PhD, FRCPCP Associate Professor of Neurology The University of Western Ontario London, Ontario, Canada A John Wiley & Sons, Ltd., Publication This edition fi rst published 2011, ® 2011 by Blackwell Publishing Ltd Blackwell Publishing was acquired by John Wiley & Sons in February 2007. Blackwell’s publishing program has been merged with Wiley’s global Scientifi c, Technical and Medical business to form Wiley-Blackwell. Registered offi ce: John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK Editorial offi ces: 9600 Garsington Road, Oxford, OX4 2DQ, UK The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK 111 River Street, Hoboken, NJ 07030-5774, USA For details of our global editorial offi ces, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com/wiley-blackwell The right of the author to be identifi ed as the author of this work has been asserted in accordance with the UK Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. -

SGCE Rabbit Pab
Leader in Biomolecular Solutions for Life Science SGCE Rabbit pAb Catalog No.: A5330 Basic Information Background Catalog No. This gene encodes the epsilon member of the sarcoglycan family. Sarcoglycans are A5330 transmembrane proteins that are components of the dystrophin-glycoprotein complex, which link the actin cytoskeleton to the extracellular matrix. Unlike other family Observed MW members which are predominantly expressed in striated muscle, the epsilon 55kDa sarcoglycan is more broadly expressed. Mutations in this gene are associated with myoclonus-dystonia syndrome. This gene is imprinted, with preferential expression from Calculated MW the paternal allele. Alternatively spliced transcript variants encoding different isoforms 49kDa/51kDa/52kDa have been found for this gene. A pseudogene associated with this gene is located on chromosome 2. Category Primary antibody Applications WB,IHC,IF Cross-Reactivity Human, Mouse Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 8910 O43556 IHC 1:50 - 1:200 Immunogen 1:50 - 1:200 IF Recombinant fusion protein containing a sequence corresponding to amino acids 1-317 of human SGCE (NP_003910.1). Synonyms SGCE;DYT11;ESG;epsilon-SG Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using SGCE antibody (A5330) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (RM00020). -

THE NON-DYSTROPHIC MYOPATHIES JOHN PEARCE, M.B., M.R.C.P., Department of Neurology, the General Infirmary, Leeds
Postgrad Med J: first published as 10.1136/pgmj.41.476.347 on 1 June 1965. Downloaded from POSTGRAD. MED. J. (1965), 41, 347 THE NON-DYSTROPHIC MYOPATHIES JOHN PEARCE, M.B., M.R.C.P., Department of Neurology, The General Infirmary, Leeds. THE TERM 'myopathy' is applied to any disorder Polymyositis may affect people of any age, of the muscle fibre, the muscle fibre membrane, but the age of onset is from 30 to 60 in 60% the myoneural junction, or the muscle connect- of cases. The chief symptom is weakness of ive tissue. 'Non-dystrophic myopathy' includes proximal muscles of the arms and/or legs all diseases of muscle excluding those genetically in every case. Distal muscles are affected in determined primary degenerative myopathies, one third, and the neck muscles in two thirds collectively known as Muscular Dystrophy. of patients. Fever, muscular pain and tenderness TABLE 1 are seen most often in the more acute forms, CLASSIFICATION OF NON-DYSTROPHIC MYOPATHIES and their absence should not lead to neglecting 1. Inflammatory Polymyositis as a Myopathy Connective tissue disorders polymyositis possible diagnosis. Other inflammatory mvopathies Acute polymyositis is not common, but may 2. Metabolic Familial periodic paralysis progress rapidly and involve the respiratory Myopathy Muscle glycogenoses muscles, sometimes with a fatal termination Myoglobinuric myopathies within a few weeks or months. Myopathies associated with Subacute and electrolyte imbalance chronic forms are more frequent, and present Unclassified myopathies with a progressive weakness and moderate of shoulder 3. Endocrine Thyrotoxicosis wasting and pelvic girdle muscles. Myopathy Cushing's Syndrome There is sometimes no systemic disturbance, Steroid myopathy and pain and tenderness are lacking. -

Periodic Paralysis
Periodic Paralysis In Focus Dear Readers Fast Facts This “In Focus” report is the third in a series of MDA’s three-year commitment for all Hypokalemic periodic paralysis MDA comprehensive reports about the latest in periodic paralysis research as of March Hypokalemic PP can begin anywhere from neuromuscular disease research and manage- 2009 is $1,938,367. The Association’s early childhood to the 30s, with periodic ment. allocation for research on hyperkalemic attacks of severe weakness lasting hours This report focuses on the periodic and hypokalemic periodic paralysis to days. The frequency of attacks gener- paralyses, a group of disorders that result from research since 1950 is $8,125,341. ally lessens in the 40s or 50s. Permanent malfunctions in so-called ion channels, micro- MDA’s allocation for the recently weakness may persist between attacks, scopic tunnels that make possible high-speed identified Andersen-Tawil syndrome usually beginning in middle age and pro- movement of electrically charged particles is $515,430 since 2001. MDA is cur- gressing slowly over years. across barriers inside cells and between cells rently funding 11 grants in the periodic The most common underlying cause and their surroundings. paralyses. is any of several genetic mutations in When ion channels fail to open or close The periodic paralyses are gener- a gene on chromosome 1 that carries according to an exquisitely fine-tuned program, ally divided into hyperkalemic periodic instructions for a calcium channel protein episodes of paralysis of the skeletal muscles paralysis, hypokalemic periodic paralysis in skeletal muscle fibers. When this chan- and even temporary irregularities in the heart- and Andersen-Tawil syndrome. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627).