The Role of the B-Type Phospholipases in S. Cerevisiae: Function, Regulation, and Physiological Relevance in Lipid Homeostasis Beth a Surlow
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Materials Supplemental Table 1
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2016 Supplemental Materials Supplemental Table 1. The differentially expressed proteins from rat pancreas identified by proteomics (SAP vs. SO) No. Protein name Gene name ratio P value 1 Metallothionein Mt1m 3.35 6.34E-07 2 Neutrophil antibiotic peptide NP-2 Defa 3.3 8.39E-07 3 Ilf2 protein Ilf2 3.18 1.75E-06 4 Numb isoform o/o rCG 3.12 2.73E-06 5 Lysozyme Lyz2 3.01 5.63E-06 6 Glucagon Gcg 2.89 1.17E-05 7 Serine protease HTRA1 Htra1 2.75 2.97E-05 8 Alpha 2 macroglobulin cardiac isoform (Fragment) 2.75 2.97E-05 9 Myosin IF (Predicted) Myo1f 2.65 5.53E-05 10 Neuroendocrine secretory protein 55 Gnas 2.61 7.60E-05 11 Matrix metallopeptidase 8 Mmp8 2.57 9.47E-05 12 Protein Tnks1bp1 Tnks1bp1 2.53 1.22E-04 13 Alpha-parvin Parva 2.47 1.78E-04 14 C4b-binding protein alpha chain C4bpa 2.42 2.53E-04 15 Protein KTI12 homolog Kti12 2.41 2.74E-04 16 Protein Rab11fip5 Rab11fip5 2.41 2.84E-04 17 Protein Mcpt1l3 Mcpt1l3 2.33 4.43E-04 18 Phospholipase B-like 1 Plbd1 2.33 4.76E-04 Aldehyde dehydrogenase (NAD), cytosolic 19 2.32 4.93E-04 (Fragments) 20 Protein Dpy19l2 Dpy19l2 2.3 5.68E-04 21 Regenerating islet-derived 3 alpha, isoform CRA_a Reg3a 2.27 6.74E-04 22 60S acidic ribosomal protein P1 Rplp1 2.26 7.22E-04 23 Serum albumin Alb 2.25 7.98E-04 24 Ribonuclease 4 Rnase4 2.24 8.25E-04 25 Cct-5 protein (Fragment) Cct5 2.24 8.52E-04 26 Protein S100-A9 S100a9 2.22 9.71E-04 27 Creatine kinase M-type Ckm 2.21 1.00E-03 28 Protein Larp4b Larp4b 2.18 1.25E-03 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Isolation and Characterization of the Prolyl Aminopeptidase Gene (Pap) from Aeromonas Sobria: Comparison with the Bacillus Coagulans Enzyme1
J. Biochem. 116, 818-825 (1994) Isolation and Characterization of the Prolyl Aminopeptidase Gene (pap) from Aeromonas sobria: Comparison with the Bacillus coagulans Enzyme1 Ana Kitazono,* Atsuko Kitano,* Daisuke Tsuru,•õ and Tadashi Yoshimoto*,2 *School of Pharmaceutical Sciences , Nagasaki University, 1-14 Bunkyo-machi, Nagasaki, Nagasaki 852; and •õ Department of Applied Microbiology, Kumamoto Institute of Technology, 4-22-1 Ikeda, Kumamoto, Kumamoto 860 Received for publication, May 16, 1994 The Aeromonas sobria pap gene encoding prolyl aminopeptidase (PAP) was cloned. It consists of 425 codons and encodes a homotetrameric enzyme of 205kDa. The purified enzyme showed an almost absolute specificity for amino-terminal proline. Proline and hydroxyproline residues from many peptide and amide substrates could be easily removed, while no activity was detected for substrates having other amino terminals. The enzyme was very similar to that from Bacillus coagulans in many aspects, such as the strong inhibition caused by PCMB and the weak or no inhibition caused by DFP and chelators, respectively. However, these enzymes show only 15% identity in their amino acid sequences. Differences were also observed in their molecular weight, stability and activity toward some peptide substrates. When aligning the deduced amino acid sequence with known sequences from other microorganisms, conserved sequences were found at the amino-terminal region; the significance of these conserved regions is discussed. Based on the results of this work, and on the studies available to date, the occurrence of at least two types of PAPs is postulated. One group would be formed by the Bacillus, Neisseria, and Lactobacillus enzymes, and the other by enzymes such as the Aeromonas PAP. -

P-Glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain and Blood Disposition of the Mtor Inhibitor Everolimus (Afinitor) in Mice
Published OnlineFirst April 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1759 Clinical Cancer Cancer Therapy: Preclinical Research P-Glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain and Blood Disposition of the mTOR Inhibitor Everolimus (Afinitor) in Mice Seng Chuan Tang1, Rolf W. Sparidans3, Ka Lei Cheung4, Tatsuki Fukami5, Selvi Durmus1, Els Wagenaar1, Tsuyoshi Yokoi5, Bart J.M. van Vlijmen4, Jos H. Beijnen2,3, and Alfred H. Schinkel1 Abstract Purpose: To clarify the role of ABCB1, ABCG2, and CYP3A in blood and brain exposure of everolimus using knockout mouse models. À À À À À À À À Experimental Design: We used wild-type, Abcb1a/1b / , Abcg2 / , Abcb1a/1b;Abcg2 / , and Cyp3a / mice to study everolimus oral bioavailability and brain accumulation. Results: Following everolimus administration, brain concentrations and brain-to-liver ratios were À À À À À À substantially increased in Abcb1a/1b / and Abcb1a/1b;Abcg2 / , but not Abcg2 / mice. The fraction of everolimus located in the plasma compartment was highly increased in all knockout strains. In vitro, everolimus was rapidly degraded in wild-type but not knockout plasma. Carboxylesterase 1c (Ces1c), a plasma carboxylesterase gene, was highly upregulated (80-fold) in the liver of knockout mice relative to wild-type mice, and plasma Ces1c likely protected everolimus from degradation by binding and stabilizing it. This binding was prevented by preincubation with the carboxylesterase inhibitor BNPP. In vivo knockdown experiments confirmed the involvement of Ces1c in everolimus stabilization. Everolimus also markedly inhibited the hydrolysis of irinotecan and p-nitrophenyl acetate by mouse plasma carboxylesterase À À and recombinant human CES2, respectively. -

Protein Network Analyses of Pulmonary Endothelial Cells In
www.nature.com/scientificreports OPEN Protein network analyses of pulmonary endothelial cells in chronic thromboembolic pulmonary hypertension Sarath Babu Nukala1,8,9*, Olga Tura‑Ceide3,4,5,9, Giancarlo Aldini1, Valérie F. E. D. Smolders2,3, Isabel Blanco3,4, Victor I. Peinado3,4, Manuel Castell6, Joan Albert Barber3,4, Alessandra Altomare1, Giovanna Baron1, Marina Carini1, Marta Cascante2,7,9 & Alfonsina D’Amato1,9* Chronic thromboembolic pulmonary hypertension (CTEPH) is a vascular disease characterized by the presence of organized thromboembolic material in pulmonary arteries leading to increased vascular resistance, heart failure and death. Dysfunction of endothelial cells is involved in CTEPH. The present study describes for the frst time the molecular processes underlying endothelial dysfunction in the development of the CTEPH. The advanced analytical approach and the protein network analyses of patient derived CTEPH endothelial cells allowed the quantitation of 3258 proteins. The 673 diferentially regulated proteins were associated with functional and disease protein network modules. The protein network analyses resulted in the characterization of dysregulated pathways associated with endothelial dysfunction, such as mitochondrial dysfunction, oxidative phosphorylation, sirtuin signaling, infammatory response, oxidative stress and fatty acid metabolism related pathways. In addition, the quantifcation of advanced oxidation protein products, total protein carbonyl content, and intracellular reactive oxygen species resulted increased -

Epistatic Interactions Associated with Fatty Acid
FACULDADE DE CIÊNCIAS AGRÁRIAS E VETERINÁRIAS UNIVERSIDADE ESTADUAL PAULISTA CÂMPUS DE JABOTICABAL EPISTATIC INTERACTIONS ASSOCIATED WITH FATTY ACID PROFILE OF BEEF FROM NELLORE CATTLE Sabrina Thaise Amorim Zootecnista 2020 FACULDADE DE CIÊNCIAS AGRÁRIAS E VETERINÁRIAS UNIVERSIDADE ESTADUAL PAULISTA CÂMPUS DE JABOTICABAL EPISTATIC INTERACTIONS ASSOCIATED WITH FATTY ACID PROFILE OF BEEF FROM NELLORE CATTLE Sabrina Thaise Amorim Orientador: Prof. Dr. Fernando Sebastián Baldi Rey Co-orientador: Dr. Fernando Brito Lopes Co-orientadora: Dra. Nedenia Bonvino Stafuzza Dissertação apresentada à Faculdade de Ciências Agrárias e Veterinárias – Unesp, Campus de Jaboticabal, como parte das exigências para a obtenção do título de Mestre em Genética e Melhoramento Animal. 2020 Amorim, Sabrina Thaise A524e Epistatic interactions associated with fatty acid profile of beef from Nellore cattle / Sabrina Thaise Amorim. -- Jaboticabal, 2020 173 p. Dissertação (mestrado) - Universidade Estadual Paulista (Unesp), Faculdade de Ciências Agrárias e Veterinárias, Jaboticabal Orientador: Fernando Sebastián Baldi Rey Coorientador: Fernando Brito Lopes 1. Genética. 2. Melhoramento Animal. 3. Genômica. I. Título. Sistema de geração automática de fichas catalográficas da Unesp. Biblioteca da Faculdade de Ciências Agrárias e Veterinárias, Jaboticabal. Dados fornecidos pelo autor(a). Essa ficha não pode ser modificada. DADOS CURRICULARES DO AUTOR Sabrina Thaise Amorim, nascida em 02 de julho de 1995 na cidade de Brusque – Santa Catarina, filha de Alexandre Adriano Amorim e Liliane RaQuel Pavesi Amorim. Iniciou em março de 2013 o curso de graduação em Zootecnia na Universidade Federal de Santa Catarina, obtendo o título de Zootecnista em dezembro de 2017. Durante a graduação foi bolsista de Iniciação Científica do CNPq, monitora da disciplina “Genética Aplicada à Zootecnia”, integrante do Grupo de Pesquisa em Produção Animal e integrante do Laboratório de Pesquisa e Ensino em Genética Animal (LEPGA) da mesma instituição de fomento, sob a orientação do Prof. -
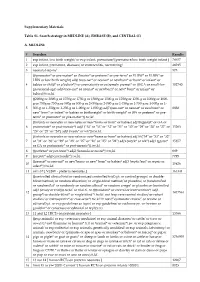
EMBASE (B), and CENTRAL (C) A. MEDLINE # Searches
Supplementary Materials Table S1. Search strategy in MEDLINE (A), EMBASE (B), and CENTRAL (C) A. MEDLINE # Searches Results 1 exp infant, low birth weight/ or exp infant, premature/ [premature/low birth weight infant ] 78657 2 exp infant, premature, diseases/ or enterocolitis, necrotizing/ 46015 3 neonatal sepsis/ 575 (((prematur* or pre-matur* or i?matur* or preterm* or pre-term* or VLBW* or ELBW* or LBW or low birth weight) adj6 (neo-nat* or neonat* or newborn* or born* or infant* or 4 babies or child* or p?ediatr*)) or prematurity or extremely premat* or ((SGA or small-for- 102740 gestational-age) adj6 (neo-nat* or neonat* or newborn* or new* born* or infant* or babies))).tw,kf. ((2000g or 2000-g or 1750g or 1750-g or 1500g or 1500-g or 1250g or 1250-g or 1000g or 1000- g or 750g or 750-g or 500g or 500-g or 2-000g or 2-000-g or 1-750g or 1-750-g or 1-500g or 1- 5 500-g or 1-250g or 1-250-g or 1-000g or 1-000-g) adj7 (neo-nat* or neonat* or newborn* or 8838 new* born* or infant* or babies or birthweight* or birth weight* or BW or preterm* or pre- term* or prematur* or pre-matur*)).tw,kf. ((infants or neonates or neo-nates or new*borns or born* or babies) adj18 (gestat* or GA or 6 postmenstr* or post-menstr*) adj3 ("34" or "33" or "32" or "31" or "30" or "29" or "28" or "27" or 15263 "26" or "25" or "24") adj3 (week* or wk*)).tw,kf. -

Thèse De Doctorat
UNIVERSITÉ PARIS-SUD ÉCOLE DOCTORALE 418 : DE CANCÉROLOGIE Laboratoire : Oncogenèse des épithéliums digestifs – Institut Cochin THÈSE DE DOCTORAT SCIENCES DE LA VIE ET DE LA SANTÉ par Pierre-Alexandre JUST Etude du rôle de LKB1 dans le foie Date de soutenance : 10/12/2014 Composition du jury : Directeur de thèse : Christine PERRET DR1, INSERM, Paris Rapporteurs : Valérie PARADIS PU-PH, Université Paris Diderot, Paris Anne-Françoise BURNOL DR1, CNRS, Paris Examinateurs : Benoît TERRIS PU-PH, Université Paris Descartes, Paris Jean ROSENBAUM DR1, INSERM, Bordeaux Président : Christian AUCLAIR PU, ENS, Cachan ii Remerciements Au moment d’achever la rédaction de ce mémoire, je souhaite remercier un grand nombre de personnes qui ont permis cette aventure dans le monde LKB1. Monsieur le professeur Christian Auclair, depuis mon Master 2, je suis étu- diant au sein de votre École doctorale et j’ai pu apprécier l’incroyable diversité, la pluridisciplinarité et la qualité de son enseignement. Vous avez à plusieurs reprises jugé mon travail lors de différentes présentations et vos remarques m’ont toujours été sources de réflexions fructueuses. Soyez-en remercié. Madame le professeur Valérie Paradis, vous me faites l’honneur de juger mon travail. Votre travail de recherche est en effet pour moi un exemple, alliant intelligemment observations anatomocliniques et confrontations biologiques. J’ap- précie votre raisonnement, partant d’approches morphologiques et aboutissant à de nouveaux paradigmes pour la carcinogenèse hépatique, notamment celle liée au syndrome métabolique. Soyez-en remerciée. Chère Anne-Françoise, voilà quelques années que je fréquente épisodique- ment ton bureau pour bénéficier de tes discussions pertinentes. Tu t’es toujours rendue disponible pour entendre mes hypothèses, souvent farfelues, et apporter, sans aucun a priori (ce qui est une qualité malheureusement trop rare dans le monde de la recherche), des éléments de réponse et des conseils expérimentaux de premier choix. -

Metabolic Regulation by Lipid Activated Receptors by Maxwell A
Metabolic Regulation by Lipid Activated Receptors By Maxwell A Ruby A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy In Molecular & Biochemical Nutrition In the Graduate Division Of the University of California, Berkeley Committee in charge: Professor Marc K. Hellerstein, Chair Professor Ronald M. Krauss Professor George A. Brooks Professor Andreas Stahl Fall 2010 Abstract Metabolic Regulation by Lipid Activated Receptors By Maxwell Alexander Ruby Doctor of Philosophy in Molecular & Biochemical Nutrition University of California, Berkeley Professor Marc K. Hellerstein, Chair Obesity and related metabolic disorders have reached epidemic levels with dire public health consequences. Efforts to stem the tide focus on behavioral and pharmacological interventions. Several hypolipidemic pharmaceutical agents target endogenous lipid receptors, including the peroxisomal proliferator activated receptor α (PPAR α) and cannabinoid receptor 1 (CB1). To further the understanding of these clinically relevant receptors, we elucidated the biochemical basis of PPAR α activation by lipoprotein lipolysis products and the metabolic and transcriptional responses to elevated endocannabinoid signaling. PPAR α is activated by fatty acids and their derivatives in vitro. While several specific pathways have been implicated in the generation of PPAR α ligands, we focused on lipoprotein lipase mediated lipolysis of triglyceride rich lipoproteins. Fatty acids activated PPAR α similarly to VLDL lipolytic products. Unbound fatty acid concentration determined the extent of PPAR α activation. Lipolysis of VLDL, but not physiological unbound fatty acid concentrations, created the fatty acid uptake necessary to stimulate PPAR α. Consistent with a role for vascular lipases in the activation of PPAR α, administration of a lipase inhibitor (p-407) prevented PPAR α dependent induction of target genes in fasted mice. -

Genetic and Molecular Analysis of Two New Loci Controlling Flowering in Garden Pea
Genetic and molecular analysis of two new loci controlling flowering in garden pea By A S M Mainul Hasan School of Natural Sciences Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy University of Tasmania, July 2018 Declaration of originality This thesis contains no material which has been accepted for a degree or diploma by the University or any other institution, except by way of background information and duly acknowledge in the thesis, and to the best of my knowledge and belief no material previously published or written by another person except where due acknowledgement is made in the text of the thesis, nor does the thesis contain any material that infringes copyright. Authority of access This thesis may be made available for loan. Copying and communication of any part of this thesis is prohibited for two years from the date this statement was signed; after that time limited copying and communication is permitted in accordance with the Copyright Act 1968. Date: 6-07-2018 A S M Mainul Hasan i Abstract Flowering is one of the key developmental process associated with the life cycle of plant and it is regulated by different environmental factors and endogenous cues. In the model species Arabidopsis thaliana a mobile protein, FLOWERING LOCUS T (FT) plays central role to mediate flowering time and expression of FT is regulated by photoperiod. While flowering mechanisms are well-understood in A. thaliana, knowledge about this process is limited in legume (family Fabaceae) which are the second major group of crops after cereals in satisfying the global demand for food and fodder. -

A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases
catalysts Review A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases Efstratios Nikolaivits 1,† , Maria Kanelli 1,†, Maria Dimarogona 2 and Evangelos Topakas 1,* 1 IndBioCat Group, Biotechnology Laboratory, School of Chemical Engineering, National Technical University of Athens, 9 Heroon Polytechniou Str., Zographou Campus, 15780 Athens, Greece; [email protected] (E.N.); [email protected] (M.K.) 2 Department of Chemical Engineering, University of Patras, 26504 Patras, Greece; [email protected] * Correspondence: [email protected]; Tel.: +30-210-772-3264 † These authors have contributed equally in this work. Received: 7 November 2018; Accepted: 27 November 2018; Published: 3 December 2018 Abstract: Cutinases are α/β hydrolases, and their role in nature is the degradation of cutin. Such enzymes are usually produced by phytopathogenic microorganisms in order to penetrate their hosts. The first focused studies on cutinases started around 50 years ago. Since then, numerous cutinases have been isolated and characterized, aiming at the elucidation of their structure–function relations. Our deeper understanding of cutinases determines the applications by which they could be utilized; from food processing and detergents, to ester synthesis and polymerizations. However, cutinases are mainly efficient in the degradation of polyesters, a natural function. Therefore, these enzymes have been successfully applied for the biodegradation of plastics, as well as for the delicate superficial hydrolysis of polymeric materials prior to their functionalization. Even though research on this family of enzymes essentially began five decades ago, they are still involved in many reports; novel enzymes are being discovered, and new fields of applications arise, leading to numerous related publications per year.