Purification and Properties of a Carboxylesterase from Germinated Finger Millet (Eleusine Coracana Gaertn.)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Isolation and Characterization of the Prolyl Aminopeptidase Gene (Pap) from Aeromonas Sobria: Comparison with the Bacillus Coagulans Enzyme1
J. Biochem. 116, 818-825 (1994) Isolation and Characterization of the Prolyl Aminopeptidase Gene (pap) from Aeromonas sobria: Comparison with the Bacillus coagulans Enzyme1 Ana Kitazono,* Atsuko Kitano,* Daisuke Tsuru,•õ and Tadashi Yoshimoto*,2 *School of Pharmaceutical Sciences , Nagasaki University, 1-14 Bunkyo-machi, Nagasaki, Nagasaki 852; and •õ Department of Applied Microbiology, Kumamoto Institute of Technology, 4-22-1 Ikeda, Kumamoto, Kumamoto 860 Received for publication, May 16, 1994 The Aeromonas sobria pap gene encoding prolyl aminopeptidase (PAP) was cloned. It consists of 425 codons and encodes a homotetrameric enzyme of 205kDa. The purified enzyme showed an almost absolute specificity for amino-terminal proline. Proline and hydroxyproline residues from many peptide and amide substrates could be easily removed, while no activity was detected for substrates having other amino terminals. The enzyme was very similar to that from Bacillus coagulans in many aspects, such as the strong inhibition caused by PCMB and the weak or no inhibition caused by DFP and chelators, respectively. However, these enzymes show only 15% identity in their amino acid sequences. Differences were also observed in their molecular weight, stability and activity toward some peptide substrates. When aligning the deduced amino acid sequence with known sequences from other microorganisms, conserved sequences were found at the amino-terminal region; the significance of these conserved regions is discussed. Based on the results of this work, and on the studies available to date, the occurrence of at least two types of PAPs is postulated. One group would be formed by the Bacillus, Neisseria, and Lactobacillus enzymes, and the other by enzymes such as the Aeromonas PAP. -

P-Glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain and Blood Disposition of the Mtor Inhibitor Everolimus (Afinitor) in Mice
Published OnlineFirst April 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1759 Clinical Cancer Cancer Therapy: Preclinical Research P-Glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain and Blood Disposition of the mTOR Inhibitor Everolimus (Afinitor) in Mice Seng Chuan Tang1, Rolf W. Sparidans3, Ka Lei Cheung4, Tatsuki Fukami5, Selvi Durmus1, Els Wagenaar1, Tsuyoshi Yokoi5, Bart J.M. van Vlijmen4, Jos H. Beijnen2,3, and Alfred H. Schinkel1 Abstract Purpose: To clarify the role of ABCB1, ABCG2, and CYP3A in blood and brain exposure of everolimus using knockout mouse models. À À À À À À À À Experimental Design: We used wild-type, Abcb1a/1b / , Abcg2 / , Abcb1a/1b;Abcg2 / , and Cyp3a / mice to study everolimus oral bioavailability and brain accumulation. Results: Following everolimus administration, brain concentrations and brain-to-liver ratios were À À À À À À substantially increased in Abcb1a/1b / and Abcb1a/1b;Abcg2 / , but not Abcg2 / mice. The fraction of everolimus located in the plasma compartment was highly increased in all knockout strains. In vitro, everolimus was rapidly degraded in wild-type but not knockout plasma. Carboxylesterase 1c (Ces1c), a plasma carboxylesterase gene, was highly upregulated (80-fold) in the liver of knockout mice relative to wild-type mice, and plasma Ces1c likely protected everolimus from degradation by binding and stabilizing it. This binding was prevented by preincubation with the carboxylesterase inhibitor BNPP. In vivo knockdown experiments confirmed the involvement of Ces1c in everolimus stabilization. Everolimus also markedly inhibited the hydrolysis of irinotecan and p-nitrophenyl acetate by mouse plasma carboxylesterase À À and recombinant human CES2, respectively. -
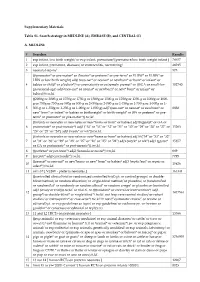
EMBASE (B), and CENTRAL (C) A. MEDLINE # Searches
Supplementary Materials Table S1. Search strategy in MEDLINE (A), EMBASE (B), and CENTRAL (C) A. MEDLINE # Searches Results 1 exp infant, low birth weight/ or exp infant, premature/ [premature/low birth weight infant ] 78657 2 exp infant, premature, diseases/ or enterocolitis, necrotizing/ 46015 3 neonatal sepsis/ 575 (((prematur* or pre-matur* or i?matur* or preterm* or pre-term* or VLBW* or ELBW* or LBW or low birth weight) adj6 (neo-nat* or neonat* or newborn* or born* or infant* or 4 babies or child* or p?ediatr*)) or prematurity or extremely premat* or ((SGA or small-for- 102740 gestational-age) adj6 (neo-nat* or neonat* or newborn* or new* born* or infant* or babies))).tw,kf. ((2000g or 2000-g or 1750g or 1750-g or 1500g or 1500-g or 1250g or 1250-g or 1000g or 1000- g or 750g or 750-g or 500g or 500-g or 2-000g or 2-000-g or 1-750g or 1-750-g or 1-500g or 1- 5 500-g or 1-250g or 1-250-g or 1-000g or 1-000-g) adj7 (neo-nat* or neonat* or newborn* or 8838 new* born* or infant* or babies or birthweight* or birth weight* or BW or preterm* or pre- term* or prematur* or pre-matur*)).tw,kf. ((infants or neonates or neo-nates or new*borns or born* or babies) adj18 (gestat* or GA or 6 postmenstr* or post-menstr*) adj3 ("34" or "33" or "32" or "31" or "30" or "29" or "28" or "27" or 15263 "26" or "25" or "24") adj3 (week* or wk*)).tw,kf. -

The Role of the B-Type Phospholipases in S. Cerevisiae: Function, Regulation, and Physiological Relevance in Lipid Homeostasis Beth a Surlow
Duquesne University Duquesne Scholarship Collection Electronic Theses and Dissertations Summer 2014 The Role of the B-Type Phospholipases in S. cerevisiae: Function, Regulation, and Physiological Relevance in Lipid Homeostasis Beth A Surlow Follow this and additional works at: https://dsc.duq.edu/etd Recommended Citation Surlow, B. (2014). The Role of the B-Type Phospholipases in S. cerevisiae: Function, Regulation, and Physiological Relevance in Lipid Homeostasis (Doctoral dissertation, Duquesne University). Retrieved from https://dsc.duq.edu/etd/1255 This Immediate Access is brought to you for free and open access by Duquesne Scholarship Collection. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Duquesne Scholarship Collection. For more information, please contact [email protected]. THE ROLE OF B-TYPE PHOSPHOLIPASES IN S. CEREVISIAE: FUNCTION, REGULATION, AND PHYSIOLOGICAL RELEVANCE IN LIPID HOMEOSTASIS A Dissertation Submitted to the Bayer School of Natural and Environmental Sciences Duquesne University In partial fulfillment of the requirements for the degree of Doctor of Philosophy By Beth A. Surlow August 2014 Copyright by Beth A. Surlow 2014 THE ROLE OF B-TYPE PHOSPHOLIPASES IN S. CEREVISIAE: FUNCTION, REGULATION, AND PHYSIOLOGICAL RELEVANCE IN LIPID HOMEOSTASIS By Beth A. Surlow Approved June 5, 2014 ________________________________ ________________________________ Dr. Jana Patton-Vogt Dr. Philip Auron Associate Professor of Biological Professor of Biological Sciences Sciences (Committee Member) (Committee Chair) ________________________________ ________________________________ Dr. Joseph McCormick Dr. Jeffrey Brodsky Chair and Associate Professor of Professor of Biological Sciences Biological Sciences University of Pittsburgh (Committee Member) (Committee Member) ________________________________ Dr. Philip Reeder Dean, Bayer School of Natural and Environmental Science iii ABSTRACT THE ROLE OF B-TYPE PHOSPHOLIPASES IN S. -

Metabolic Regulation by Lipid Activated Receptors by Maxwell A
Metabolic Regulation by Lipid Activated Receptors By Maxwell A Ruby A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy In Molecular & Biochemical Nutrition In the Graduate Division Of the University of California, Berkeley Committee in charge: Professor Marc K. Hellerstein, Chair Professor Ronald M. Krauss Professor George A. Brooks Professor Andreas Stahl Fall 2010 Abstract Metabolic Regulation by Lipid Activated Receptors By Maxwell Alexander Ruby Doctor of Philosophy in Molecular & Biochemical Nutrition University of California, Berkeley Professor Marc K. Hellerstein, Chair Obesity and related metabolic disorders have reached epidemic levels with dire public health consequences. Efforts to stem the tide focus on behavioral and pharmacological interventions. Several hypolipidemic pharmaceutical agents target endogenous lipid receptors, including the peroxisomal proliferator activated receptor α (PPAR α) and cannabinoid receptor 1 (CB1). To further the understanding of these clinically relevant receptors, we elucidated the biochemical basis of PPAR α activation by lipoprotein lipolysis products and the metabolic and transcriptional responses to elevated endocannabinoid signaling. PPAR α is activated by fatty acids and their derivatives in vitro. While several specific pathways have been implicated in the generation of PPAR α ligands, we focused on lipoprotein lipase mediated lipolysis of triglyceride rich lipoproteins. Fatty acids activated PPAR α similarly to VLDL lipolytic products. Unbound fatty acid concentration determined the extent of PPAR α activation. Lipolysis of VLDL, but not physiological unbound fatty acid concentrations, created the fatty acid uptake necessary to stimulate PPAR α. Consistent with a role for vascular lipases in the activation of PPAR α, administration of a lipase inhibitor (p-407) prevented PPAR α dependent induction of target genes in fasted mice. -

Genetic and Molecular Analysis of Two New Loci Controlling Flowering in Garden Pea
Genetic and molecular analysis of two new loci controlling flowering in garden pea By A S M Mainul Hasan School of Natural Sciences Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy University of Tasmania, July 2018 Declaration of originality This thesis contains no material which has been accepted for a degree or diploma by the University or any other institution, except by way of background information and duly acknowledge in the thesis, and to the best of my knowledge and belief no material previously published or written by another person except where due acknowledgement is made in the text of the thesis, nor does the thesis contain any material that infringes copyright. Authority of access This thesis may be made available for loan. Copying and communication of any part of this thesis is prohibited for two years from the date this statement was signed; after that time limited copying and communication is permitted in accordance with the Copyright Act 1968. Date: 6-07-2018 A S M Mainul Hasan i Abstract Flowering is one of the key developmental process associated with the life cycle of plant and it is regulated by different environmental factors and endogenous cues. In the model species Arabidopsis thaliana a mobile protein, FLOWERING LOCUS T (FT) plays central role to mediate flowering time and expression of FT is regulated by photoperiod. While flowering mechanisms are well-understood in A. thaliana, knowledge about this process is limited in legume (family Fabaceae) which are the second major group of crops after cereals in satisfying the global demand for food and fodder. -

A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases
catalysts Review A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases Efstratios Nikolaivits 1,† , Maria Kanelli 1,†, Maria Dimarogona 2 and Evangelos Topakas 1,* 1 IndBioCat Group, Biotechnology Laboratory, School of Chemical Engineering, National Technical University of Athens, 9 Heroon Polytechniou Str., Zographou Campus, 15780 Athens, Greece; [email protected] (E.N.); [email protected] (M.K.) 2 Department of Chemical Engineering, University of Patras, 26504 Patras, Greece; [email protected] * Correspondence: [email protected]; Tel.: +30-210-772-3264 † These authors have contributed equally in this work. Received: 7 November 2018; Accepted: 27 November 2018; Published: 3 December 2018 Abstract: Cutinases are α/β hydrolases, and their role in nature is the degradation of cutin. Such enzymes are usually produced by phytopathogenic microorganisms in order to penetrate their hosts. The first focused studies on cutinases started around 50 years ago. Since then, numerous cutinases have been isolated and characterized, aiming at the elucidation of their structure–function relations. Our deeper understanding of cutinases determines the applications by which they could be utilized; from food processing and detergents, to ester synthesis and polymerizations. However, cutinases are mainly efficient in the degradation of polyesters, a natural function. Therefore, these enzymes have been successfully applied for the biodegradation of plastics, as well as for the delicate superficial hydrolysis of polymeric materials prior to their functionalization. Even though research on this family of enzymes essentially began five decades ago, they are still involved in many reports; novel enzymes are being discovered, and new fields of applications arise, leading to numerous related publications per year. -

Table SI. Downregulated Differentially Expressed Genes Identified in Lung Adenocarcinomas
Table SI. Downregulated differentially expressed genes identified in lung adenocarcinomas. Gene symbol Gene name HEG1 Heart development protein with EGF like domains 1 LIMS2 LIM zinc finger domain containing 2 GRK5 G protein‑coupled receptor kinase 5 TNXB///TNXA Tenascin XB///tenascin XA (pseudogene) OLFML1 Olfactomedin like 1 HSPA12B Heat shock protein family A (Hsp70) member 12B CYYR1 Cysteine and tyrosine rich 1 CCDC102B Coiled‑coil domain containing 102B DOK6 Docking protein 6 GIMAP1 GTPase, IMAP family member 1 LIMCH1 LIM and calponin homology domains 1 NEBL Nebulette WWC2///CLDN22 WW and C2 domain containing 2///claudin 22 DACH1 Dachshund family transcription factor 1 PTPN21 Protein tyrosine phosphatase, non‑receptor type 21 SYNM Synemin SLC39A8 Solute carrier family 39 member 8 RECK Reversion inducing cysteine rich protein with kazal motifs EMP2 Epithelial membrane protein 2 AFAP1L1 Actin filament associated protein 1 like 1 NEXN Nexilin F‑actin binding protein ITM2A Integral membrane protein 2A SMAD6 SMAD family member 6 LAMP3 Lysosomal associated membrane protein 3 GNLY Granulysin QKI QKI, KH domain containing RNA binding PKNOX2 PBX/knotted 1 homeobox 2 FPR2 Formyl peptide receptor 2 SH2D3C SH2 domain containing 3C SH3GL3 SH3 domain containing GRB2 like 3, endophilin A3 SHANK3 SH3 and multiple ankyrin repeat domains 3 ECSCR Endothelial cell surface expressed chemotaxis and apoptosis regulator TMEM204 Transmembrane protein 204 TRHDE Thyrotropin releasing hormone degrading enzyme KIAA1462 KIAA1462 ITIH5 Inter‑alpha‑trypsin inhibitor -

Over-Expression and Characterization of a Glyoxalase 2 Like Enzyme
MIAMI UNIVERSITY The Graduate School Certificate for Approving the Dissertation We hereby approve the Dissertation of Pattraranee Limphong Candidate for the Degree: Doctor of Philosophy _______________________________________________ Director Dr. Christopher A. Makaroff _______________________________________________ Reader Dr. Michael W. Crowder _______________________________________________ Reader Dr. John W. Hawes _______________________________________________ Reader Dr. Neil D. Danielson _______________________________________________ Graduate School Representative Dr. Qingshun Li ABSTRACT OVER-EXPRESSION AND CHARACTERIZATION OF A GLYOXALASE 2 LIKE ENZYME By Pattraranee Limphong This dissertation consists of seven chapters to better probe and understand the structure and function of a glyoxalase 2 (GLX2) like enzyme from Arabidopsis thaliana and human. Chapter 1 provides a general background of the glyoxalase system followed by the physiological roles of the glyoxalase system, and inhibitors of the glyoxalase enzymes, glyoxalase 1 and glyoxalase 2. Chapter 2 focuses on over-expression and characterization of GLX2-1 (putative glyoxalase 2, isozyme1) from Arabidopsis thaliana. Metal analyses, kinetics, and spectroscopic studies suggested that GLX2-1 contains a dinuclear metal binding site but is not a glyoxalase 2. Chapter 3 investigates the physiological role of GLX2-1. A commercial survey substrate system and β-lactamase substrates were used. The results showed that GLX2-1 exhibits β-lactamase activity. Chapter 4 presents the results of experiments to convert inactive GLX2-1 into an active GLX2 enzyme. Substrate binding residues were mutated at positions 225, 253, 255, 332, and 335 in GLX2-1. The results showed that the R253H GLX2-1 does hydrolyze S-D-lactoylglutathione (SLG) when the substrate binding ligands were generated on the R253H GLX2-1 enzyme. Chapter 5 provides detailed structural information on the human GLX2 metal center and insights concerning the structure and kinetic mechanism of the enzyme. -

Carboxylesterase 1 Plays an Essential Role in Non
CARBOXYLESTERASE 1 PLAYS A PROTECTIVE ROLE AGAINST METABOLIC DISEASE A dissertation submitted to Kent State University in partial fulfillment of the requirements for the degree of Doctor of Philosophy By Jiesi Xu May 2016 © Copyright All rights reserved Except for previously published materials Dissertation written by Jiesi Xu B.S., Jilin University 2008 Ph.D., Kent State University 2016 Approved by ___________________________________ , Chair, Doctoral Dissertation Committee Dr. Yanqiao Zhang, M.D., Associate Professor, NEOMED ___________________________________ , Member, Doctoral Dissertation Committee Dr. John Y.L. Chiang Ph.D., Distinguished Professor, NEOMED ___________________________________, Member, Doctoral Dissertation Committee Dr. Colleen M. Novak, Ph.D., Associate Professor, Kent State University ___________________________________, Member, Doctoral Dissertation Committee Dr. Min You, Ph.D., Professor, NEOMED ___________________________________, Member, Doctoral Dissertation Committee Dr. Eric M. Mintz, Ph.D., Professor, Associate dean, Kent State University Accepted by ___________________________________ , Director, School of Biomedical Sciences Dr. Ernest Freeman, Ph.D. ___________________________________ , Dean, College of Arts and Sciences Dr. James L. Blank, Ph.D ii TABLE OF CONTENTS TABLE OF CONTENTS LIST OF FIGURES ............................................................................................................v LIST OF ABBREVIATIONS .........................................................................................viii -

(12) United States Patent (10) Patent No.: US 8,561,811 B2 Bluchel Et Al
USOO8561811 B2 (12) United States Patent (10) Patent No.: US 8,561,811 B2 Bluchel et al. (45) Date of Patent: Oct. 22, 2013 (54) SUBSTRATE FOR IMMOBILIZING (56) References Cited FUNCTIONAL SUBSTANCES AND METHOD FOR PREPARING THE SAME U.S. PATENT DOCUMENTS 3,952,053 A 4, 1976 Brown, Jr. et al. (71) Applicants: Christian Gert Bluchel, Singapore 4.415,663 A 1 1/1983 Symon et al. (SG); Yanmei Wang, Singapore (SG) 4,576,928 A 3, 1986 Tani et al. 4.915,839 A 4, 1990 Marinaccio et al. (72) Inventors: Christian Gert Bluchel, Singapore 6,946,527 B2 9, 2005 Lemke et al. (SG); Yanmei Wang, Singapore (SG) FOREIGN PATENT DOCUMENTS (73) Assignee: Temasek Polytechnic, Singapore (SG) CN 101596422 A 12/2009 JP 2253813 A 10, 1990 (*) Notice: Subject to any disclaimer, the term of this JP 2258006 A 10, 1990 patent is extended or adjusted under 35 WO O2O2585 A2 1, 2002 U.S.C. 154(b) by 0 days. OTHER PUBLICATIONS (21) Appl. No.: 13/837,254 Inaternational Search Report for PCT/SG2011/000069 mailing date (22) Filed: Mar 15, 2013 of Apr. 12, 2011. Suen, Shing-Yi, et al. “Comparison of Ligand Density and Protein (65) Prior Publication Data Adsorption on Dye Affinity Membranes Using Difference Spacer Arms'. Separation Science and Technology, 35:1 (2000), pp. 69-87. US 2013/0210111A1 Aug. 15, 2013 Related U.S. Application Data Primary Examiner — Chester Barry (62) Division of application No. 13/580,055, filed as (74) Attorney, Agent, or Firm — Cantor Colburn LLP application No.